An in-depth review of column chromatography for protein purification and survey result from formal publications.
Purified proteins are required for many experimental applications, including structural studies and in vitro biochemical assays. Proteins can be obtained from a tissue or, more often, by their overexpression in a model organism, such as bacteria, yeast, or mammalian cells in culture. Protein purification involves isolating proteins from the source, based on differences in their physical properties. The objective of a protein purification scheme is to retain the largest amount of the functional protein with fewest contaminants. The purification scheme of a protein must be optimized to complete this process in the least number of steps.

The article reviews four types of column chromatography that are commonly used in protein purification and discusses the advantages, disadvantages, and potential problems of each. This article also reports a Labome survey on 98 publications. The survey indicates that affinity column chromatography, mainly that based on HIS, GST, and FLAG tags, and size exclusion chromatography are the main methods cited in the publications. GE Healthcare is the major supplier of reagents and instruments used in protein purification.
The most important consideration in the development of a protein purification scheme is the downstream application of the purified protein. Both the quantity and purity of the protein must be sufficient for experimental analysis. Additionally, information about the behavior of the protein must be taken into consideration, as well-folded and functional protein is required for downstream studies. During purification and subsequent storage, many processes can occur that affect protein quality: protein unfolding, aggregation, degradation, and loss of function. Careful planning to purify protein as quickly as possible and under the most stabilizing conditions will maximize the chance of a successful purification scheme.

The solution conditions of a protein at each step of the purification scheme are essential in maintaining protein stability and function. Proteins should be kept in a well-buffered environment to prevent sudden changes in pH that could irreversibly affect their folding, solubility, and function.
A buffer is a solution containing a conjugate acid/base pair. The pH range of a buffer is based on its pKa, defined as the pH at which 50% of the molecules are in their acidic form, and 50% are in their basic form (Figure 1). A general rule regarding buffers is that the pH of the buffer solution should be within 1.0 pH unit of the pKa to provide appropriate buffering capacity. This ensures that there is a sufficient amount of the molecule in both its acidic and basic forms to neutralize the solution in case of H+ or OH- influx. Thus, buffers prevent pH changes that could negatively affect protein stability.

A good buffer must exhibit the following characteristics [1] :
- Water solubility
- Chemical stability
- High buffering capacity at desired pH
- Compatibility with analytical and experimental applications
- Compatibility with other solution components
Many components can serve as biological buffers. The most commonly used buffering components have a near neutral pKa, as they can be used at a physiological pH. Four of the most common biological buffers are listed in Table 1, along with the pH range at which they can be used, and advantages and disadvantages that might affect their usage in protein purification. Typically, these buffers are used at concentrations above 25mM to ensure adequate buffering capacity.
| Buffer | pH range | Advantages and Disadvantages |
|---|---|---|
| Phosphate | 5.8-8.0 | pH is not dependent on temperature Inexpensive Transparent in the UV range Cannot be used with divalent cations Some proteins may precipitate with sodium phosphate buffer, potassium phosphate buffer can be used. |
| MOPS | 6.5-7.9 | Cannot be autoclaved pH is somewhat dependent on temperature High buffering capacity at physiological pH |
| HEPES | 6.8-8.2 | Cannot be autoclaved pH is somewhat dependent on temperature Can form radicals under certain conditions [2] |
| Tris | 7.5-9.0 | pH is dependent on temperature and dilution Inexpensive Can interfere with the activity of some enzymes [3], [4] Transparent in the UV range |
In addition to an appropriate buffering system, solutions used during protein purification from lysis to storage often contain many other components that play a role in facilitating protein purity, stability, and function.
Protease inhibitors are often added to the lysis buffer and in early steps of the purification scheme to prevent degradation of the target protein by endogenous proteases. These are generally not needed toward later stages of the purification, as most or all of the contaminating proteases have been separated from the protein of interest. Metal chelating reagents, such as EDTA or EGTA, are often added to the storage buffer. These metal chelators bind to Mg2+ and, thus, prevent cleavage of the purified protein by contaminating metalloproteases. Other additives are often used to protect proteins against damage and enhance their solubility.
| Type | Function | Commonly Used Reagents |
|---|---|---|
| Reducing Agents | Protect against oxidative damage | 2-mercaptoethanol (BME) Dithiothreitol (DTT) Tris (2-carboxyethyl) phosphine (TCEP) |
| Protease Inhibitors | Inhibit endogenous proteases from degrading proteins | Leupeptin (serine and cysteine protease inhibitor) Pepstatin A (aspartic acid protease inhibitor) PMSF (serine protease inhibitor) |
| Metal Chelators | Inactivate metalloproteases | EDTA EGTA |
| Osmolytes | Stabilize protein structure and enhance solubility | Glycerol Detergents (e.g., CHAPS, NP-40, Triton X-100, DDM) Sugars (e.g., glucose, sucrose) |
| Ionic Stabilizers | Enhance solubility | Salts (e.g., NaCl, KCl, (NH4)2SO4 |
Additives should only be used if necessary. Trial and error are often required to determine the specific additives that are beneficial to a particular protein purification scheme.
Other factors also contribute to protein stability during a purification scheme. The least manipulation of a protein during its purification is always the best. Designing a purification scheme that uses the minimum number of steps in the shortest amount of time ensures the highest yield of functional protein. Additionally, it is often best to keep the protein cold throughout the purification. Typically, purification is performed at 4°C, as this lower temperature both slows down the rate of proteolysis (in the event of contaminating proteases) and promotes structural integrity of proteins.
Before one can proceed to purify the protein of interest an initial crude sample must be prepared. The first consideration, which takes place well in advance of performing the actual purification, is the source of the protein of interest. This could be a native source such as liver, muscle or brain tissue, though in the post-genomic area it is now relatively rare for investigators to purify proteins from native sources. However, there is still sometimes the need if the investigator wishes to link a catalytic activity to a specific protein sequence.
Nowadays it is far more common for proteins to be purified from recombinant sources. Important decisions need to be made in advance to optimize the subsequent purification. The investigator needs to consider the end-use of the protein (e.g., enzyme assay, structural studies, antibody generation) as this will dictate the quantities and purity required of the final protein preparation. An important consideration is the choice of expression system (see Labome article Recombinant Protein Expression: Vector-Host Systems) for a discussion of the most widely used expression systems based on an unbiased survey of the literature. While a detailed discussion of protein expression is outside the scope of this article a few basic points are worth considering at this point as they directly impact upon the subsequent purification of the protein.
Which expression system will give the highest level of expression? As a general rule, the higher the expression level, the easier it will be to obtain large quantities of highly purified protein.
If opting for E.coli expression, is the ultimate aim soluble expression or insoluble expression in the form of inclusions bodies? Isolation of inclusion bodies can in itself constitute a significant purification step due to the high abundance of the recombinant protein within inclusion bodies; this has to be balanced against the ease/difficulty of solubilizing and subsequently refolding the target protein from the inclusion bodies and the ultimate yield of the soluble protein [5]. Much work has been done to optimize the yields of functional protein from inclusion bodies [6].
Another consideration is whether to target intracellular or extracellular (secreted) expression. Intracellular expression requires the protein to be purified away from a significant number of host cellular proteins. By contrast, efficient secretion of the target protein can result in the protein having to be purified away from a small number of secreted host proteins, especially if the host cells are grown under serum-free conditions [7].
Regardless of the source of the target protein, the initial preparation of a crude sample as a starting point for purification is important and needs to be considered at the same time as thinking about the expression and purification strategies.
Extracellular secretion of the target protein may allow for a rapid and straightforward affinity purification protocol to be used [7] ; the only sample preparation required may be the decanting of the conditioned media from adherent cells or low-speed centrifugation to remove suspension cells. Of course, it may be necessary to add protease inhibitors or adjust the pH in preparation for the first chromatographic step, but these are simple steps to carry out. However, if ion-exchange is to be performed as the first purification step the sample may first need to be desalted. This can be technically challenging if large volumes of a conditioned medium are to be processed; dialysis or cross-flow filtration are often used depending on the volume of medium to be desalted.
If the target protein is expressed intracellularly, the cells first need to be harvested by centrifugation before being resuspended in an appropriate lysis buffer. As discussed above, the lysis buffer needs to contain an appropriate buffer and other additives to ensure maximum stability of the target protein. The composition of the sample/lysis buffer also needs to be compatible with the subsequent purification step(s) if the investigator is to avoid time-consuming buffer exchange steps before column chromatography.
Next, an efficient method is required for lysing the cells. Various methods have been described in the literature. E .coli cells can be lysed by French press (though this method is not readily scalable), sonication or detergent-based lysis (many commercial lysis reagents are available). The efficiency of detergent-based lysis can be improved by the addition of lysozyme to the lysis buffer. Detergent-based lysis is very mild and does not result in significant shearing of bacterial DNA. Thus the inclusion of DNAase preparations (highly pure preparations are commercially available) is usually required to reduce sample viscosity and produce a sample with good flow characteristics [8].
Sonication or detergent-based methods can also lyse mammalian and insect cells. DNAase treatment may be required to reduce sample viscosity if there is significant lysis of the nuclear membrane [8].
Once the cells (microbial/insect/mammalian) have been lysed it is usually necessary to remove cellular debris by centrifugation (typically 15 000 x g for 15 minutes at 4oC to avoid clogging of chromatography columns [8]. The resulting supernatant is the starting point for column chromatographic purification of the target protein.
The principle of column chromatography is to separate a large pool of proteins into many smaller pools, some of which are enriched in the protein of interest. While expensive and specialized equipment is available for column chromatography, only basic equipment is required.

Basic equipment for column chromatography:
- Stationary phase: an inert matrix, often with an attached functional group to facilitate protein interaction, used to separate proteins. The choice of stationary phase and functional group depends on both the type of chromatography that is being performed and the method by which it will be carried out.
- Column: a cylindrical, glass reservoir available in various lengths and diameters. Columns can be purchased pre-packed with a stationary phase and ready to attach to automated chromatography systems (discussed in Part 2 of this section) or can be bought empty for manual packing. Different types of columns are required for automated versus gravity flow chromatography.
- Solvents: buffers containing additives used for equilibrating, washing, and eluting proteins from the stationary phase. Different types of chromatography require different solvent conditions.
- Collection tubes: vessels for collecting elution samples. Special tubes are required for automated fraction collectors; however, any tube or vessel is appropriate for manual fraction collection.
- Assay to measure purity: an approach for determining the relative amount of a specific protein to the total protein in a sample. The fractions containing the protein of interest must be determined after each step before proceeding to the next step in the purification scheme. Some common methods of analyzing purity will be discussed in a later section.
Column chromatography can be performed with automated systems (Figure 2), which use a pump to force solvent over a packed column at a set flow rate, or can be run by gravity flow. Both automated and gravity flow systems can be coupled to automatic fraction collecting systems. There are advantages and disadvantages to each system. GE Healthcare AKTA FPLC system is a very common choice [9-18].
| Type of System | Advantages | Disadvantages |
|---|---|---|
| Automated | Can let run by itself Often coupled to an absorbance detector Can program equilibration and wash steps Easy to set up a gradient for elution Very reproducible | Requires specific, costly equipment The maximum flow rate is dependent on the pressure limit of the column |
| Gravity Flow | Less expensive The user has more control Can make adjustments during run | More labor intensive The flow rate is limited by gravity |
The four major types of column chromatography include affinity chromatography, ion exchange chromatography (IEX), hydrophobic interaction chromatography (HIC), and size exclusion chromatography (SEC). Most purification schemes require the use of more than one of these types of chromatographic procedures to yield the necessary purity for downstream applications. Choosing the most appropriate chromatographic method(s) and the order of these methods is essential in optimizing a protein purification scheme.
| Type of Chromatography | Separates Proteins By | Bind With | Elute With |
|---|---|---|---|
| Affinity | A specific interaction | No competing ligand | Competing ligand (specific); conditions that disrupt protein/protein interactions (non-specific) |
| Ion Exchange | Net surface charge | Low ionic strength | High ionic strength; Increased (cation exchange) or decreased (anion exchange) pH |
| Hydrophobic Interaction | Hydrophobicity | High ionic strength | Low ionic strength |
| Size Exclusion | Hydrodynamic radii |
By analyzing the sequence of a protein, unique characteristics can be identified that might assist in its purification. The size and charge of a protein (at a specific pH) can be determined, along with the identification of large stretches of hydrophobic residues.
Affinity chromatography relies on the specific and reversible binding of a protein to a matrix-bound ligand. The ligand can bind directly to either the protein of interest, for example, cAMP-resin for cAMP-binding PKA RIα and RIIβ proteins [19], or a tag that is covalently attached to the protein. Affinity chromatography is often the most robust purification procedure and is typically used in the early stages of the purification scheme. Depending on the downstream application, affinity purification might be the only chromatographic step required to achieve adequate purity.

The stationary phase for affinity chromatography is made of an inert matrix covalently attached to a ligand that specifically binds to a protein or group of proteins. The inert matrix is typically composed of cross-linked agarose or polyacrylamide. Proteins can be purified by affinity chromatography in a selective or non-selective manner. In selective affinity chromatography, a ligand specific for a protein or a covalently attached tag is used. In non-selective affinity chromatography such as Protein A, G, L for immunoglobulin, or heparin for DNA-binding proteins, or lectin for glycoproteins, the ligand binds to a group of proteins with similar binding capabilities. For example, SARS-CoV-2 S envelope protein expressed to produce a COVID 19 vaccine is purified through lentil lectin affinity column chromatography [20].
In both types of affinity chromatography, proteins are loaded on the column under conditions that influence binding between the protein (or tag) and its ligand. The bound protein is washed under conditions that do not disrupt the specific interaction, but that can disrupt any non-specific interactions between contaminating proteins and the stationary phase. The bound protein is then eluted with a buffer containing a competing molecule or conditions that disrupt all protein/protein interactions. Competing molecules bind to the ligand, displacing the protein of interest. This competing molecule is typically removed from the protein of interest either through another chromatographic procedure or dialysis. Methods for eluting proteins from the stationary phase by disrupting all protein/protein interactions include adjusting the pH or ionic strength of the buffer. These methods can affect protein stability, and it is suggested that the eluted protein be immediately neutralized or diluted to minimize protein damage. For some forms of affinity chromatography, alternative elution conditions have been described to maximize the yield of functional proteins [21, 22].
| Protein to Purify | Ligand | Elute With |
|---|---|---|
| Antibody (antigen-specific) | Antigenic peptide | Free peptide |
| Polyhistidine-tagged protein | Ni2+ or Co2+ | Imidazole or free histidine |
| FLAG-tagged protein | FLAG-specific antibody | FLAG peptide or low pH |
| GST-tagged protein | Reduced glutathione | Free glutathione |
| Myc-tagged protein | Myc-specific antibody | Low pH |
| Antibody (class-specific) | Protein A , G, or L or protamine | Extremes in pH |
| DNA-binding protein | Heparin | High ionic strength |
In designing a plasmid for protein expression, affinity tags can be inserted on either the N- or C-terminus (or in rare cases, in flexible loops of proteins with known structures) to assist in purification. See Labome survey on protein/protide tags for a list of common tags and tag-related discussions.
Antibodies are often purified based on the highly specific interaction that occurs between an antibody and its antigen (the sequence recognized by the antibody). A peptide containing the antigen can be coupled to a matrix for specific binding of the antibody. Lowering the pH of the elution buffer disrupts the antibody/peptide interaction to release the bound antibody. This method is often used commercially in the isolation of antibodies from crude serum.

Proteins can also be affinity purified in a non-selective manner. In non-selective purification, the ligand attached to the stationary phase binds to a group of proteins with similar binding partners. An example of non-selective affinity purification is in the purification of DNA-binding proteins. Heparin mimics DNA both in its structure and its charge and can be used as the ligand for the affinity purification of DNA-binding proteins. While all DNA-binding proteins could theoretically bind to this stationary phase, most other proteins will flow through without binding, leading to sufficient enrichment of the protein of interest. Another example is the enrichment of antibodies by the binding of their constant (Fc) region to the ligand, Protein A, G, or L. GE Healthcare protein A affinity columns are a common choice [23-25] and also from other suppliers [26], as is protein G [27] or protein L [28], which binds the V kappa light chain variable region. For IgM, protamine affinity chromatography can be used [29].
Using a similar mode of binding (an antibody bound to a Protein A, G, or L ligand), the antigen-binding (Fab) region of the antibody is still available for binding to its specific antigen. Thus, particular proteins can be purified based on the specific interaction between the coupled ligand/antibody and the antibody/antigen.
Common problems and troubleshooting are listed in Table 6.
| Problem | Cause | Solution |
|---|---|---|
| Protein does not bind | Tag was not translated or is not accessible | Check plasmid sequence or move the tag to a different location |
| Binding conditions are not appropriate | Adjust buffer conditions | |
| Not enough time was allowed for binding | Decrease flow rate or stop column to allow incubation | |
| Protein does not elute | Affinity between ligand and tag is very high | Increase concentration of competitor (specific) or stringency of conditions (non-specific) |
| Protein aggregated on column | Adjust buffer conditions for more protein stability | |
| Low resolution | Flow rate is either too fast or too slow | Adjust flow rate |
| The column was not washed sufficiently | Wash with higher stringency buffer; Clean stationary phase according to manufacturer | |
| Protein aggregated on column | Adjust buffer conditions for more protein stability | |
| Elution conditions are not stringent enough | Increase concentration of competitor (specific) or adjust conditions (non-specific) | |
| Protein loses activity during procedure | Protein is unfolded or aggregated | Adjust buffer conditions for more protein stability |
| A cofactor required for activity was removed during purification | Add cofactor |
Ion exchange chromatography (IEX) separates proteins based on their net surface charge, through electrostatic interactions that occur between proteins and a charged stationary phase. Two types of IEX exist (1) anion exchange (a positively charged stationary phase that binds to negatively charged proteins); and (2) cation exchange (a negatively charged stationary phase that binds to positively charged proteins). Ion exchange chromatography is commonly used as an intermediate step in a protein purification scheme; however, it can yield high resolution for some proteins when used earlier or later during the purification.
All proteins exhibit a net charge that depends on the amino acid composition of the protein and any covalently attached modifications. The net charge of a protein is influenced by the pH of the solvent that it is dissolved in, as solvents exchange hydrogen ions with proteins. The isoelectric point (pI) of a protein is the pH at which the protein has no net charge. At a pH above the pI, a protein will have a net negative charge, while a pH below the pI will lead to a net positive charge. Thus, the pH of the solvent can be adjusted to facilitate binding to IEX or to promote elution of a bound protein.

Theoretically, all proteins can bind to both cation exchange and anion exchange if the pH of the buffer is adjusted accordingly. However, for protein purification, the stability of the protein is the most important consideration in choosing purification conditions and, thus, the most appropriate column for protein binding. Therefore, it is necessary to determine whether conditions required for binding to either form of ion exchange chromatography affect protein stability and function. Typically, conditions for binding to one type of ion exchange is more suitable for a particular protein than the other.
Knowledge of a protein's isoelectric point can aid in determining the most appropriate type of ion exchange chromatography. Online tools are available for calculating the theoretical pI of a protein EXPASY. These calculations are based entirely on the amino acid sequence of a protein and do not take the three-dimensional structure of the protein into consideration. In its native state, some residues of a protein are more exposed than others and, thus, the actual pI and net surface charge is sometimes different from those determined theoretically [30]. The distribution of amino acids relative to one another can also affect a protein's pI [31], and this is not taken into consideration with theoretical pI calculations. Some techniques allow for the experimental determination of a protein's pI in its native state, including electrophoretic isoelectric focusing [32], capillary isoelectric focusing [33], and a more recent high-throughput luminescence-based approach [34].
Typically, binding of a protein to IEX must be determined by trial and error, using solvents with a range of pH values, to determine the optimum pH for protein retention. A solvent pH that is about one pH unit away from the pI is usually sufficient for protein binding [35] ; however, in some cases, a pH further from the pI is required [36].
The stationary phase of IEX is composed of an inert agarose-based or polymeric matrix with a covalently bound charged group. Matrix particles are available in many sizes and can either be non-porous or can contain pores of variable size. The choice of the matrix depends on the desired binding capacity, resolution, and flow rate. Smaller particle sizes offer higher resolution, but require lower flow rates and take longer to run. Porous matrices offer a higher binding capacity than non-porous matrices. In a non-porous matrix, proteins cannot enter the resin and, thus, offer increased sample recovery, greater resolution, and shorter run times than porous matrices. One study showed that for most proteins, resolution, and recovery are similar when separated by a porous versus non-porous matrix; however, for larger proteins, porous matrices cause a loss in resolution based on size-exclusion effects [37].

The four charged functional groups most commonly used in IEX are shown below. These are classified as either strong or weak exchangers, with strong exchangers being ionized over a wider pH range than weak exchangers.
Anion exchange (positively charged stationary phase):
- quaternary ammonium (Q) - strong anion exchanger
- diethylaminoethyl (DEAE) - weak anion exchanger
Cation exchange (negatively charged stationary phase):
- methyl sulfonate (S) - strong cation exchanger
- carboxymethyl (CM) - weak cation exchanger
Strong exchangers should be used when the pH required for binding is very acidic or basic (assuming this pH is also appropriate for maintaining protein stability), as the functional groups remain charged over a larger pH range, [38]. Weak exchangers have been shown to provide less retention of some proteins, enabling their elution at lower ionic strength [39]. Because high ionic strength can affect the stability of some proteins [40], weak exchangers might be more suitable for proteins that do not require an extreme pH for binding.
In ion exchange chromatography, the stationary phase is first equilibrated with a low ionic strength buffer. The protein sample is then loaded onto the stationary phase in the same low ionic strength buffer used for equilibration. The bound protein is washed extensively before elution with a higher ionic strength buffer or, in some cases, a buffer with altered pH (Figure 6). Counterions in the elution buffer interact with the charged stationary phase, displacing the bound protein. Certain salts are more efficient for use in ion exchange chromatography than others, due to their ability to displace bound proteins and their effects on protein stability [41]. Typically NaCl or KCl is used for elution, with Na+ or K+ serving as the countercation in cation exchange chromatography and Cl- serving as the counteranion in anion exchange chromatography. Alternatively, the pH of the buffer can be altered to reduce the protein's charge and disrupt its interaction with the stationary phase. For proteins bound to a cation exchange stationary phase, increasing the buffer pH will lead to a lesser positive charge on the protein, and subsequent elution from the column. For proteins bound to an anion exchange stationary phase, decreasing the buffer pH will lead to a lesser negative charge on the protein, and subsequent elution from the column.
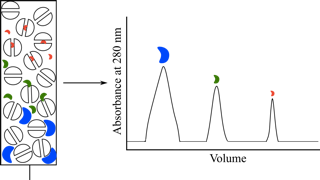
Alternatively, the pH of the buffer can be adjusted so that the protein of interest does not bind to the ion exchange stationary phase while contaminating proteins do. In this case, the protein of interest is collected in the flow through, while some contaminating proteins are removed by their binding to the stationary phase.
As with other chromatographic methods, ion exchange chromatography may require some troubleshooting to determine the optimum conditions for protein binding, protein elution, and adequate resolution. In some cases, a pure protein may exhibit multiple chromatographic peaks due to the presence of more than one conformations [42].
| Problem | Cause | Solution |
|---|---|---|
| Protein does not bind | Column was not equilibrated | Run more equilibration buffer through the column and reload protein |
| The ionic strength of binding buffer is too high | Lower ionic strength of the buffer | |
| pH is not far enough from pI | Adjust buffer pH (lower for cation exchange, higher for anion exchange) | |
| Protein does not elute | Ionic strength of elution buffer is too low | Increase ionic strength |
| Protein aggregated on column | Adjust buffer conditions for more protein stability | |
| Low resolution | Flow rate is either too fast or too slow | Adjust flow rate |
| The column was not washed sufficiently | Wash with a higher ionic strength buffer; Clean stationary phase according to manufacturer | |
| Protein aggregated on column | Adjust buffer conditions for more protein stability | |
| Protein loses activity during procedure | Protein is unfolded or aggregated | Adjust buffer conditions for more protein stability |
| A cofactor required for activity was removed during purification | Add cofactor |
Hydrophobic interaction chromatography (HIC) separates proteins based on their hydrophobicity and is often used as an intermediate step in a purification scheme. Proteins are bound to a stationary phase in a high ionic strength buffer and, thus, HIC can typically be performed immediately after ion exchange chromatography with no buffer exchange or dilution required. HIC is also commonly performed after an ammonium sulfate precipitation, a procedure that can be used to remove proteins by precipitating some quickly, but not all, proteins with salt. HIC is sometimes applicable in early steps of a purification scheme or as a final step in the removal of trace impurities from the protein of interest.
The presence of salt ions in solution can lead to the partial unfolding of a protein and the exposure of normally buried hydrophobic residues. Proteins that bind to the stationary phase adopt their native conformation as a buffer with a lower ionic strength is added. This decreases the exposure of hydrophobic residues that can interact with the stationary phase and facilitates elution from the column. For proteins that can refold spontaneously through a decrease in ionic strength, HIC can be a valuable chromatographic method for purification.
Typically, the ionic strength of the binding buffer should be as low as possible to bind the protein of interest, while preventing its precipitation. If the ionic strength required for protein binding is high and causes precipitation of the protein of interest, a lower ionic strength can be used. In this case, the chromatography procedure can be used to separate all of the binding proteins from the protein of interest that flows through without binding.
Before loading the sample on the column, the stationary phase must be equilibrated with the high ionic strength buffer (the same buffer that the protein sample will be applied in). The sample is then loaded onto the column, the column is washed extensively, and the protein is eluted with a low ionic strength buffer (Figures 7 and 8).
The stationary phase used in hydrophobic interaction chromatography is composed of a base matrix of either cross-linked agarose or a synthetic copolymer. An alkyl or aryl ligand is then conjugated to the base matrix, providing specificity for hydrophobic molecules.
Types of functional group:
- Alkyl - a hydrocarbon chain of various length; often a butyl or octyl group is used. The binding capacity of the stationary phase is increased with increasing alkyl chain length [43]. The functional groups bind proteins based entirely on the hydrophobicity of the protein
- Aryl - a functional group derived from an aromatic ring; often a phenyl group. Aryl groups offer increasing specificity, as proteins can also interact with the functional group through base stacking interactions.
The type and concentration of salt used for binding and elution must be determined empirically for each protein. Additionally, the refolding and function of the protein must be ensured after its elution.
Like other forms of column chromatography, an optimum HIC procedure can require a great deal of troubleshooting. Adjusting buffer conditions and stationary phase are required for each protein to ensure optimal separation.
| Problem | Cause | Solution |
|---|---|---|
| Protein does not bind | Column was not equilibrated | Run more equilibration buffer through the column and reload protein |
| The ionic strength of binding buffer is too low | Increase ionic strength of the buffer | |
| Protein aggregated at the ionic strength used | Decrease ionic strength of buffer or try a different salt | |
| Protein does not elute | Ionic strength of elution buffer is too high | Decrease ionic strength |
| Protein aggregated on column | Adjust buffer conditions for more protein stability | |
| Retention is too high | Try a different stationary phase that offers less retention | |
| Low resolution | Flow rate is either too fast or too slow | Adjust flow rate |
| Column was not washed sufficiently | Wash with a lower ionic strength buffer; Clean stationary phase according to manufacturer | |
| Protein aggregated on column | Adjust buffer conditions for more protein stability | |
| Poor retention on column | Try a different stationary phase that offers greater retention | |
| Protein loses activity during procedure | Protein is unfolded or aggregated | Adjust buffer conditions for more protein stability |
| Protein did not return to its native conformation | Try binding with a different salt or at a lower ionic strength; Include additives for protein stability | |
| A cofactor required for activity was removed during purification | Add cofactor |
Size exclusion chromatography separates proteins by their hydrodynamic radius, a property determined by both the size and shape of the molecule. Unlike the other chromatographic procedures described previously, proteins do not bind to a stationary phase in the SEC. Instead, proteins are separated by the speed at which they navigate through an inert stationary phase (Figure 9).
SEC is a valuable method most often used in the final steps of a purification scheme due to its ability to differentiate between different species of a protein. Oligomeric species [44], unfolded protein molecules [45], and truncated species [46] can all be separated from the native protein, while simultaneously exchanging buffer components. Often, SEC is used as a faster and more reliable method of buffer exchange than dialysis, as it is compatible with more solvents and requires less buffer. A single solvent is used throughout the entire SEC procedure, and commercially available SEC stationary phases are compatible with most commonly used buffer components.
The type of stationary phase used and the length of the column both play critical roles in influencing the resolution of proteins separated by SEC. Several types of stationary phases are commercially available, and the best option depends on both the molecular weight of the proteins requiring separation and the conditions under which the separation will be performed.
| Type of Stationary Phase | Matrix | Features |
|---|---|---|
| Sephadex | Cross-linked dextran and epichlorohydrin | Offers quick buffer exchange and group separation Works well for molecular weight determination Autoclavable The matrix can shrink in certain solvents Specific types of Sephadex are available for use with organic solvents |
| Sephacryl | Cross-linked allyl dextran and N, N -methylene bisacrylamide | Separates molecules over a large molecular weight range Autoclavable Works with aqueous and organic solvents High recovery |
| Sepharose | Cross-linked agarose | Separates molecules over a large molecular weight range High recovery Cannot be autoclaved |
| Superose | Highly cross-linked agarose | Works with aqueous and organic solvents Autoclavable Hydrophobic interactions between proteins and matrix are possible Compatible with viscous solvents |
| Superdex | Cross-linked agarose and dextran | Works with aqueous and organic solvents Autoclavable High resolution High recovery |
Superdex is often the best choice for typical SEC procedures, as it is compatible with most solvents and offers the highest resolution of all the available stationary phases.
As with other chromatographic methods, there are advantages and disadvantages to SEC, and the decision to use this method depends on both the protein and its downstream application.
Advantages of SEC:
- Provides buffer exchange, and desalting.
- Enables the separation of similar species (e.g., truncations and oligomers) that might not be separated by other purification techniques.
- Is compatible with many solvents.
- Does not depend on any specific protein property for retention and elution.
Disadvantages of SEC:
- Performance is very sensitive to column packing.
- Can have nonspecific interactions between protein and resin, which decreases resolution.
- Offers low resolution for complex protein mixtures.
- The sample must be loaded at a small volume for adequate resolution. This can be problematic for proteins that precipitate at high concentrations.
Optimizing SEC conditions to achieve maximum resolution is often time-consuming, but certain factors can have a huge impact on resolution.
Conditions that increase protein resolution with SEC:
- Minimize the sample volume. The smaller the volume, the less diffuse the eluted fractions will be.
- Add salt to the buffer. A small amount of salt will help prevent nonspecific interactions between the protein and stationary phase. This will allow all protein molecules to run consistently over the length of the column.
- Use a moderate flow rate. Running a column too quickly will not allow time for small molecules to enter the matrix pores, while a flow rate that is too slow allows more time for sample diffusion.
- Ensure that sample and solvent viscosities are similar. Adjust sample conditions to approximately match that of the running buffer.
- Adjust the column length. A column that is too short will not allow adequate protein separation. Alternatively, a column that is too long will lead to diffusion of the protein sample.
- Re-pack the column. Column packing can have a huge effect on protein resolution.
If particles are not well-dispersed or if air bubbles are trapped in the column, navigation through the stationary phase will not occur properly. Additionally, if the column is allowed to run dry, it must be repacked. Poor column packing is often to blame for unexplained low resolution.
Because SEC does not separate proteins based on interactions with a functional group, all proteins are eluted under the same conditions and, thus, resolution can only be obtained for proteins of very different hydrodynamic radii. Therefore, SEC is not an appropriate chromatographic method for the initial stages of a purification scheme when there are many contaminating proteins. SEC does, however, offer a quick and reliable method for removing salts or small molecules from a sample between early or intermediate stages of the purification. In the final stages of purification, when only trace contaminants exist, SEC is a valuable method for protein isolation and exchange into a storage buffer.
Proteins that bind to a stationary phase are eluted with solvent conditions that disrupt the binding interactions. These elution conditions vary both by type of chromatography and properties of the protein of interest. There are three general methods of protein elution: batchwise elution, stepwise elution, and linear gradient elution. The best method to choose depends on the type of chromatography being performed and the required resolution.
- Batchwise - a single elution condition displaces all bound proteins in a single step. This mechanism works best for chromatographic procedures based on a very specific interaction (i.e., affinity chromatography). Batchwise elution does not offer any resolution, but it is ideal for getting rid of contaminants very quickly. It requires prior knowledge of buffer conditions required for displacement of the protein of interest.
- Stepwise - multiple batchwise elutions are performed sequentially, with more stringent conditions in each step. In stepwise elution, the number of fractions collected is dependent upon the number of sequential elution conditions. Stepwise elution provides better resolution than batchwise elution, but poorer resolution than linear gradient elution.
- Linear Gradient - multiple consecutive fractions are collected while elution conditions are adjusted linearly. A linear gradient offers the highest resolution for ion exchange chromatography and hydrophobic interaction chromatography. Typically, a large number of consecutive fractions are collected.
Size exclusion chromatography does not require any of these elution methods, as no interactions occur between the protein and the stationary phase. Protein is loaded onto the column, and a large number of fractions are collected until all proteins are eluted, without altering buffer conditions throughout the procedure.
Ideally, the elution buffer of a column is compatible with the subsequent column, eliminating the need for buffer exchange or dialysis between purification steps.
Hydroxyapatite (hydroxylated calcium phosphate) is a protein purification technique described initially in the mid-1950s [47]. The commercial availability of spherical hydroxyapatite has made hydroxyapatite column chromatography an accessible technique [48]. The mechanisms underlying the interaction of proteins with hydroxyapatite columns is complex. Proteins are most commonly eluted with phosphate gradients. (see [48] for a discussion). Hydroxyapatite has different selectivity properties to other chromatography techniques and thus can provide a useful addition to the purification of difficult to purify proteins or as a final ‘polishing’ step. Hydroxyapatite chromatography has been used successfully in the purification of therapeutic grade antibodies [49].
Chromatofocusing is a column chromatographic method that separates proteins based on their pI. Proteins are bound to specialized ion-exchange media and eluted with a pH gradient. The selectivity of chromatofocusing is different from that of other techniques and thus is useful as a final ‘polishing’ step [50].
After each chromatographic separation, fractions must be analyzed to determine the fractions that contain the protein of interest and the relative purity of each of those fractions. This analysis is necessary after each step to decide which fractions should be pooled for subsequent use. To determine purity, an assay is required that can measure the amount of a specific protein relative to the amount of total protein. The following assays are routinely used for purity analysis:
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) - a denaturing gel that separates proteins based on size. Commercially available stains allow for a visual representation of all proteins in the sample, offering a qualitative assessment of protein purity (Figure 10). SDS-PAGE is not ideal for high-throughput analysis of fractions and can take several hours; however, it is most often used because it is easy, inexpensive, and suitable for any protein.
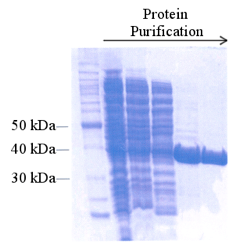 Figure 10. SDS-PAGE of samples collecting during a protein purification scheme. The gel is stained for the visualization of all proteins. From http://www.omicsonline.org/ .
Figure 10. SDS-PAGE of samples collecting during a protein purification scheme. The gel is stained for the visualization of all proteins. From http://www.omicsonline.org/ .- Spectroscopy - a method for analyzing optical properties of proteins. This technique for analyzing protein purity is only suited for proteins, such as cytochrome P450s, that have a unique spectroscopic feature. Proteins in this family absorb light at a wavelength where other proteins do not (around 420 nanometers [51] ), so a comparison of absorbance at 420 nanometers versus 280 nanometers (the wavelength at which all proteins absorb), can provide a quantitative measure of protein purity. This method is fast and high-throughput, but only suitable for some proteins.
- Protein Activity - an enzymatic test that depends on the protein of interest. This method of assessing protein purity is often coupled with another form of protein concentration determination to calculate activity relative to total protein concentration. Activity assays are only suitable for proteins with the activity that can easily be monitored in a high-throughput format, such as proteases. For some proteins, activity assays provide a fast and reliable method for protein detection. Activity measurement is often ideal for enzymes because protein that has lost activity can be excluded from subsequent use.

When proteins are deemed pure enough for use in experimental studies, they should be stored appropriately. The selection of a final storage buffer is just as important as the selection of buffers used during the purification scheme and should depend on the stability of the protein and conditions required for the downstream application of the purified protein. Often, size exclusion chromatography is selected as a final step in the purification scheme, as the storage buffer can be used in this chromatographic step to exchange the buffer effectively. The pure fractions can be pooled for immediate storage. Alternatively, the final pooled fractions can be dialyzed into the selected buffer before storage.
Protein storage conditions depend on the protein of interest and should be optimized, so the protein maintains structural and functional stability over long periods of storage. Additives are often included in the storage buffer to enhance the lifetime of purified proteins under storage conditions, and trial and error are often required to determine optimum conditions, as every protein behaves differently.
In this post-genomics era, there is much interest in high throughput purification of proteins both for structural determination and for drug discovery/compound screening purposes. To screen a wide range of constructs and select the optimal one(s) for the desired end use investigators have utilized a range of commercially available automated/robotic systems that enable the rapid, parallel, (semi-) automated purification of affinity-tagged proteins [52, 53]. The basic principles of protein still apply; liquid handling robotics / automated platforms are simply used to enable to streamline and accelerate the purification process.
Some 20 - 30% of the proteins produced by cells are integral membrane proteins, and some 50% of small molecule drugs act on membrane proteins [54]. Thus, there is great interest in solving the structure of membrane proteins. A crucial step in the purification of integral membrane proteins is their solubilization from the lipid bilayer while retaining their functional integrity. The typical approach involves the isolation of intracellular membranes by centrifugation followed by detergent solubilization of integral membrane proteins and high-speed centrifugation to remove insoluble membrane residues [55-57]. A wide range of detergents have been employed for membrane protein solubilization [58-60] and, in the absence of any literature or laboratory precedent, the investigator will need to determine the best detergent for their particular protein empirically. The solubilized membrane protein may then be purified by column chromatography in essentially the same way as for soluble proteins. However, the purification buffers will need to contain detergents to maintain the protein in a soluble state [61, 62]. Purification of membrane proteins is often extremely challenging due to loss of protein functional integrity and aggregation following initial removal from the lipid bilayer and through the various purification steps. Nonetheless, several groups have successfully purified membrane proteins in sufficient quantities for structure determination (see for example, Structural Genomics Consortium). Recently, nanodiscs have been successfully employed in the affinity purification of a Family B GPCR (see Labome article on nanodiscs) [63].
Labome surveyed publications citing protein purification methods. Table 10 lists the major suppliers and purification methods. Affinity and size exclusion methods are the most commonly used approaches in the literature. GE Healthcare is the predominant supplier for all methods, and Qiagen is the significant provider of HIS tag-based protein purification, and MilliporeSigma, of FLAG-based tag.
| type | supplier | major brand | num | sample references | |
|---|---|---|---|---|---|
| affinity | |||||
| HIS | Qiagen | Ni-NTA agarose/resin | 63 | [64, 65] | |
| GE Healthcare | Ni Sepharose, HisTrap FF/HP | 37 | [66, 67] | ||
| Clontech | TALON metal affinity | 23 | [68, 69] | ||
| Thermo Fisher / Invitrogen | HisPur Cobalt/Ni-NTA resin | 9 | [70, 71] | ||
| Roche/ MilliporeSigma | cOmplete His-Tag Purification Resins | 1 | [72] | ||
| Cube Biotech | Ni-NTA resin | 1 | [73] | ||
| GST | GE Healthcare | glutathione Sepharose 4B | 66 | [66, 74] | |
| MilliporeSigma | glutathione agarose | 3 | [75, 76] | ||
| FLAG | MilliporeSigma | FLAG M2 affinity | 17 | [77, 78] | |
| DNA binding protein | GE Healthcare | heparin | 16 | [67] | |
| maltose-binding protein / MBP | New England Biolabs | amylose resin | 10 | [73, 79] | |
| Other cited affinity-based protein purification systems include Strep-tactin resin from IBA Lifesciences for SBP-fused proteins [73], Streptactin Sepharose® High Performance from GE Healthcare (28-9355-99) [80]. | |||||
| size exclusion | |||||
| GE-Healthcare | Superdex / Superose / Sephacryl | 104 | [81] | ||
| ion exchange | |||||
| anion | GE Healthcare | Mono Q column | 11 | [79, 82] | |
| GE Healthcare | HiTrap | 9 | [67, 79, 80] | ||
| GE Healthcare | Source 15Q | 4 | [82, 83] | ||
| GE Healthcare | Q-Sepharose | 1 | [84] | ||
| Whatman | DE52 anion exchange resin | 1 | [85] | ||
| MilliporeSigma | DE52 anion exchange column | 1 | [86] | ||
| cation | GE Healthcare | Mono S column | 6 | [87] | |
| TOROH | TSKgelSP-5PW column | 1 | [82] | ||
| GE Healthcare | Source 15S | 1 | [82] | ||
| hydrophobic interaction | |||||
| GE healthcare | phenyl sepharose CL-4B | 1 | [88] | ||
| MilliporeSigma | phenyl sepharose | 1 | [84] | ||
Among the literature surveyed, protein samples were prepared through MilliporeSigma BugBuster [89, 90] or AVESTIN EmulsiFlex C-3 cell disruptor [91, 92], and were concentrated through MilliporeSigma Amicon Ultra concentrators [93, 94], Thermo Fisher Sartorius spin columns [95] or through Vivaspin concentrators [96-98]. Maini Rekdal V et al purified E. faecalis MMH594 tyrosine decarboxylase from E. coli BL21(DE3) cultures through Avestin emulsiflex C3 cell disruptor, Thermo Fisher Scientific HisPur Ni-NTA resin, and VMR spin columns ( catalog # 97027-9) [71].
- Good N, Winget G, Winter W, Connolly T, Izawa S, Singh R. Hydrogen ion buffers for biological research. Biochemistry. 1966;5:467-77 pubmed
- Grady J, Chasteen N, Harris D. Radicals from "Good's" buffers. Anal Biochem. 1988;173:111-5 pubmed
- Desmarais W, Bienvenue D, Bzymek K, Holz R, Petsko G, Ringe D. The 1.20 A resolution crystal structure of the aminopeptidase from Aeromonas proteolytica complexed with tris: a tale of buffer inhibition. Structure. 2002;10:1063-72 pubmed
- Ghalanbor Z, Ghaemi N, Marashi S, Amanlou M, Habibi Rezaei M, Khajeh K, et al. Binding of Tris to Bacillus licheniformis alpha-amylase can affect its starch hydrolysis activity. Protein Pept Lett. 2008;15:212-4 pubmed
- Scott M, Modha S, Rhodes A, Broadway N, Hardwicke P, Zhao H, et al. Efficient expression of secreted proteases via recombinant BacMam virus. Protein Expr Purif. 2007;52:104-16 pubmed
- Arakawa T, Philo J, Tsumoto K, Yumioka R, Ejima D. Elution of antibodies from a Protein-A column by aqueous arginine solutions. Protein Expr Purif. 2004;36:244-8 pubmed
- Chong S, Mersha F, Comb D, Scott M, Landry D, Vence L, et al. Single-column purification of free recombinant proteins using a self-cleavable affinity tag derived from a protein splicing element. Gene. 1997;192:271-81 pubmed
- Wichmann A, Borg H. Purification of human immunoglobulin M by affinity chromatography on protamine-Sepharose. Biochim Biophys Acta. 1977;490:363-9 pubmed
- Salaman M, Williamson A. Isoelectric focusing of proteins in the native and denatured states. Anomalous behaviour of plasma albumin. Biochem J. 1971;122:93-9 pubmed
- Vesterberg O. Isoelectric fractionation, analysis, and characterization of ampholytes in natural pH gradients. V. Separation of myoglobins and studies on their electro-chemical differences. Acta Chem Scand. 1967;21:206-16 pubmed
- Awdeh Z, Williamson A, Askonas B. Isoelectric focusing in polyacrylamide gel and its application to immunoglobulins. Nature. 1968;219:66-7 pubmed
- Righetti P. Determination of the isoelectric point of proteins by capillary isoelectric focusing. J Chromatogr A. 2004;1037:491-9 pubmed
- Ahamed T, Chilamkurthi S, Nfor B, Verhaert P, van Dedem G, van der Wielen L, et al. Selection of pH-related parameters in ion-exchange chromatography using pH-gradient operations. J Chromatogr A. 2008;1194:22-9 pubmed
- Trodler P, Nieveler J, Rusnak M, Schmid R, Pleiss J. Rational design of a new one-step purification strategy for Candida antarctica lipase B by ion-exchange chromatography. J Chromatogr A. 2008;1179:161-7 pubmed
- Duncan J, Chen A, Siebert C. Performance evaluation of non-porous versus porous ion-exchange packings in the separation of proteins by high-performance liquid chromatography. J Chromatogr. 1987;397:3-12 pubmed
- Staby A, Jensen R, Bensch M, Hubbuch J, Dünweber D, Krarup J, et al. Comparison of chromatographic ion-exchange resins VI. Weak anion-exchange resins. J Chromatogr A. 2007;1164:82-94 pubmed
- DePhillips P, Lenhoff A. Determinants of protein retention characteristics on cation-exchange adsorbents. J Chromatogr A. 2001;933:57-72 pubmed
- von Hippel P, Wong K. On the conformational stability of globular proteins. The effects of various electrolytes and nonelectrolytes on the thermal ribonuclease transition. J Biol Chem. 1965;240:3909-23 pubmed
- Tsumoto K, Ejima D, Senczuk A, Kita Y, Arakawa T. Effects of salts on protein-surface interactions: applications for column chromatography. J Pharm Sci. 2007;96:1677-90 pubmed
- Er el Z, Zaidenzaig Y, Shaltiel S. Hydrocarbon-coated sepharoses. Use in the purification of glycogen phosphorylase. Biochem Biophys Res Commun. 1972;49:383-90 pubmed
- Woodbury R, Hardy S, Randall L. Complex behavior in solution of homodimeric SecA. Protein Sci. 2002;11:875-82 pubmed
- Corbett R, Roche R. Use of high-speed size-exclusion chromatography for the study of protein folding and stability. Biochemistry. 1984;23:1888-94 pubmed
- Kim T, Paik S, Yang C. Structural and functional implications of C-terminal regions of alpha-synuclein. Biochemistry. 2002;41:13782-90 pubmed
- Hjerten S, Levin O, Tiselius A. Protein chromatography on calcium phosphate columns. Arch Biochem Biophys. 1956;65:132-55 pubmed
- Giri L. Chromatofocusing. Methods Enzymol. 1990;182:380-92 pubmed
- Schenkman J, Jansson I. Spectral analyses of cytochromes P450. Methods Mol Biol. 2006;320:11-8 pubmed
- SGC | Integral Membrane Proteins. Available from: www.thesgc.org/science/imp
- Smith S. Strategies for the Purification of Membrane Proteins. Methods Mol Biol. 2017;1485:389-400 pubmed
- Materials and Methods [ISSN : 2329-5139] is a unique online journal with regularly updated review articles on laboratory materials and methods. If you are interested in contributing a manuscript or suggesting a topic, please leave us feedback.
- reagentmethod
- Incorporating Unnatural Amino Acids into Recombinant Proteins in Living Cells
- Nanodiscs: Membrane Protein Research in Near-Native Conditions
- Protein Companies
- Protein Expression
- Protein Modification
- Protein/Peptide Tags
- Protein Quantitation
- Receptor-Ligand Binding Assays
- siRNAs and shRNAs: Tools for Protein Knockdown by Gene Silencing