The article provides a summary of protein modifications from the UniProt database and an overview of research methods for major protein modifications: phosphorylation, acetylation and methylation, ubiquitination, glycosylation, and sumoylation.
The human proteome consists of significantly more functional polypeptides than the collection of genes in the genome, due, in part, to co- and post-translational protein modifications (Fig 1A). One of the important areas of proteomic research is to identify those proteins that are post-translationally modified and their modification sites and to characterize the function of the modifications and the interaction of the modified protein within a functional cellular network.

The curated SWISS-Prot Protein Knowledgebase lists 695 unique modifications [6], as of September 24, 2022 (UniProt Release 2022_05). RESID database [7] by Dr. John S. Garavelli also provides comprehensive information for each modification.
Multiple methods to assay protein modifications have been developed over the past decades. Here we focus on the mass spectrometry as a general approach for detecting protein modifications, and specific methods for phosphorylation, ubiquitination, glycosylation, and sumoylation, as well as for analyzing histone acetylation and methylation in the context of chromatin remodeling (Table 1 and Fig 1B). Detailed protocols are not covered. Discussion about citrullination, using histone citrullination as an example, can be found at the review about histone modifications. Protein citrullination can also be investigated through site-specific incorporation of citrulline into proteins with a modified leucyl tRNA synthetase-tRNA pair [8]. The biotin-switch technique can be used to assay the degree of S-nitrosylation of protein of interest [9].
| Modification | Function | Assay |
|---|---|---|
| Phosphorylation | Intracellular signaling | Mass spectrometry Radiometry Western blotting Phos-tag electrophoresis |
| Ubiquitination | Protein degradation | Mass spectrometry In vitro ubiquitination assays Western blotting TUBEs |
| Acetylation and methylation | Chromatin regulation Transcriptional regulation | Mass spectrometry CHIP CHIP-on-CHIP |
| Glycosylation | Extracellular signaling | Mass spectrometry HPLC Electrophoresis |
| SUMOylation | Intracellular transport Transcriptional regulation Apoptosis Protein stability Stress response Cell-cycle regulation | Mass spectrometry SUBEs PRISM Western blotting [10] |
A major preliminary approach for identifying novel modification sites is the in silico analysis. Computer programs that identify putative modification sites based on sequence have been extensively utilized and are beyond the scope of this review. See Liu and Li, 2011 [11] for an excellent overview of computer programs.
Over the past 20 years, mass spectrometric analysis has become an essential tool in determining the types and sites of protein modifications. Mass spectrometric analysis can be performed on both purified proteins or a mixture of proteins, for example, cell lysates [12, 13] or tissue extracts [14, 15].
A mass spectrometer generates gas-phase ions from a sample, separates them according to their mass-to-charge ratio (m/z) and generates a record of their abundance. Mass spectrometry can be used for molecular weight determination of peptides and proteins, for determining the amino acid sequence of peptides, and for the detection of post-translational modifications, as well as the relative quantification of peptides and proteins. This method cannot be used for absolute quantification.
For the preparation of a sample for mass spectrometric analysis, the protein or lysate is digested into small peptide fragments using trypsin or, more recently, other proteases [16]. The fragments are then vaporized and analyzed to determine their m/z value. Since the sites of restriction digest are known, a computer program then determines the unique amino acid sequence of each peptide based on mass. The molecular weight and charge of a moiety such as a phosphate group are also known, the phosphorylation of specific amino acids within a peptide fragment can also be assessed.
A digested protein sample can be analyzed by matrix-assisted laser desorption/ionization (MALDI) peptide mapping or nanoelectrospray mass spectrometer. The use of these methods, however, can result in incomplete coverage, with some of the peptides clearly visible and others – not at all. This is often a major problem in analyzing complex mixtures, and for modified peptides and in the analysis of post-translational modifications, peptides are first separated by reversed-phase chromatography, with subsequent fraction collection and analysis by mass spectrometry (a method known as LC/MS) (Fig 2B) [17]. To more accurately determine the nature of the peptide modification, tandem mass spectrometry (MS/MS) experiments are often performed (Fig 2A). After the first MS step, the peptide ions collide with an inert gas, which leads to further fragmentation. These peptide fragments are then analyzed in the second MS step [18]. Some modified peptides will remain intact during this process, generating a fragmentation pattern similar to the unmodified peptide, while others will fragment significantly, generating a pattern that can provide further information about the nature and location of the modified amino acids. For example, Liu Y et al identified demethylation and methylation on differet residues of ALKBH5 with nano-ultra-performance liquid chromatography-electrospray inonization tandem mass spectrometry using Q-Exactive MS from Thermo Fisher Scientific, after immunoprecipitation and SDS-PAGE separation [19]. Hoxhaj G et al used LC-MS/MS to determine the sites on NAD kinase phosphorylated by akt kinase [20]. Black MH et al used LC-MS/MS to identify glutamylated peptides in SdeA by the Legionella meta-effector SidJ [21]. Bhogaraju S et al performed further analysis on the glutamylation of SdeA [22].
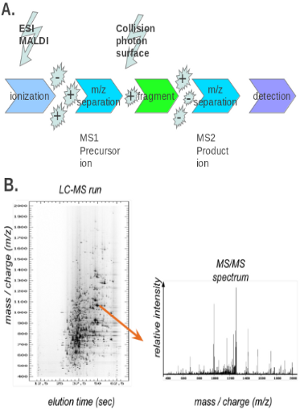
In addition to being used to determine the modification state of a single protein, mass spectrometry has been used to generate extensive datasets of proteins that carry a specific modification [23]. Some examples are Asn deamidation [14], phosphorylation [24], sumoylation [25], ubiquitination [26], non-histone [27] and histone methylation [28], glycosylation [29] or oxidized proteins [30-33]. This analysis usually involves affinity selection prior to the mass spectrometric analysis [34]. Such techniques are used to define ‘sub-proteomes’ while analyzing the activation state of entire signaling networks in cancer [35], neurodegenerative diseases [36, 37] or pathogen-host interactions [38, 39].
However, mass spectrometric analysis is costly and requires specific equipment and training. Because more quantitative data is often required, other biochemical methods are used in conjunction with mass spectrometry to analyze post-translational modifications. Moreover, MS-based analysis of PTMs has its own caveats that can lead to erroneous conclusions. Older [12], [13], and more recent [40-42] extensive reviews of the multiple MS methods, common errors and suggestions for accurate data collection and computational analysis are available [43, 44].
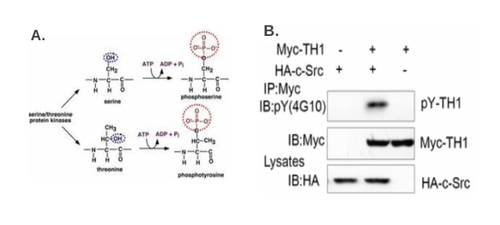
Phosphorylation, or the addition of a phosphate group to serine, threonine, or tyrosine residues, is one of most common forms of protein modification (Fig 3A). Protein phosphorylation plays an important role in intracellular signaling cascades and is reversible, fine-tuning the signal [45]. Several in vivo and in vitro methods are used to assess protein phosphorylation status, to assess phosphorylation of a specific amino acid, and to ascertain whether a specific kinase (or phosphatase) modifies a protein in question. In addition to the methods discussed below, Pro-Q® Diamond phosphoprotein gel stain can directly label phosphoproteins on a 2D PAGE gel, as in the case of SY5Y cells treated with Ab42 peptides [46].
Radiometric kinase reactions using 32P-gamma-ATP can be used to assay phosphorylation status in vitro [47]. This is also the gold standard for assessing the role of specific kinases. Purified target protein, kinase, and 32P-gamma-ATP are incubated in a reaction buffer. The reaction solution is then passed through a filter to bind protein but wash away unbound ATP, and the level of phosphorylation (radioisotope retained on a filter) is detected by a scintillation counter. Alternatively, the reaction can be resolved by SDS-PAGE and visualized by exposure to X-ray film [48]. This method is less quantitative but can confirm the molecular weight of the phosphorylated protein.
Similarly, the reaction can include phosphatase instead of the kinase to determine whether a phosphorylated protein is a substrate of a specific phosphatase.
To assay phosphorylation in vivo, radioactive pulse labeling have been utilized [49]. Cells are grown in the presence 32P-orthophosphate. The protein of interest is then immunoprecipitated with a specific antibody and the retained radioisotope quantified by scintillation counter or resolved on an SDS-PAGE and exposed to X-ray film. This method can be used to assay phosphorylation under various physiological conditions. Furthermore, in conjunction with a protein knock-down step, this method can also be used to implicate a specific kinase or phosphatase in the modification status of the target protein.
There are two categories of phospho-specific antibodies. One category is the generic phosphotyrosine, phosphoserine, and phosphothreonine antibody, which will bind to any phospho-tyrosine, phospho-serine, and phospho-threonine moieties regardless of the neighboring amino acid residues. The second category includes the antibodies (or other affinity reagents [50] ) against phospho-specific amino acid epitopes (see a summary of phospho-specific antibodies in Labome product database).
Following identification of a (putative) phosphorylation site by an in silico method or by mass spectrometry, commercially available antibodies for the general class of phosphorylation sites can be used in conjunction with an immunoprecipitation step to determine if the protein of interest is phosphorylated at a specific type of site (for example a canonical cyclin/cdk site) [51]. Antibodies generated against a specific phosphorylated amino acid, such as phosphotyrosine, can also be used (Fig 3B). This can be followed up by generating a phospho-specific antibody to the specific epitope and assaying phosphorylation by simple Western blotting. To analyze a large number of samples, an ELISA can be performed, where the substrate is captured by the membrane and detected by the phospho-specific antibody [52]. Kenner LR et al confirmed phosphorylation of human eIF2alpha through western bloting with an eIF2alpha S51 phosphorylation-specific antibody from Cell Signaling (cat# 9721) [53]. However, the specificify of phosphorylation-specific antibodies is often questioned [54].
Anti-phosphoprotein antibody microarrays are powerful tools for investigating protein phosphorylation-mediated signaling systems. These microarrays facilitate the semiquantitative measurement of along the phosphorylation status of protein kinases and their substrates. A detailed protocol is reviewed here [55].
Phos-tag is a selective phosphate-binding molecule, 1,3-bis[bis(pyridin-2-ylmethyl) amino]propan-2-olato dizinc(II), developed for MS-based proteomics [56], and can be used for separation of phosphorylated proteins in acrylamide and agarose gels [57]. By using Phos-tag SDS-PAGE various phosphorylated and unphosphorylated protein species can be visualized due to their noticeable mobility shifts [58-60]. The method is easy to use as it resembles the classical SDS-PAGE analysis. Biotinylated Phos-tag can also be used to facilitate Western blot analysis of samples [61, 62]. Protocols for Phos-tag SDS-PAGE with either alkaline pH [63] or neutral pH [64] buffer systems are now available. A review on recent advances of the Phos-tag based methodologies was published in 2015 [63]. Kenner LR et al confirmed phosphorylation of human eIF2alpha through Phos-Tag SDS-PAGE [53].
Apart from being useful tools for the identification of phosphoproteins, the Phos-tag SDS-PAGE technique, in combination with other non-denaturing gel electrophoresis methods proved to be powerful methods for the characterization of the phosphorylation-dependent activity of certain enzymes. For example, Meisrimler et al combined the Phos-tag technology with two-dimensional electrophoresis in which either non-reducing isoelectric focusing (IEF) PAGE or non-equilibrium pH gel electrophoresis (NEPHGE) was used in the first dimension and high resolution clear native electrophoresis (hrCNE) in the second dimension, to investigate phosphorylation-dependent changes in enzyme activity [65]. The authors discussed in detail the advantages and the disadvantages of these new techniques [65]. Kitchen P et al used Phos-tag reagent from NARD Chemicals, Kobe City, Japan to analyze the phosphorylation of AQP4 [66].
Protein ubiquitination, or the covalent attachment of ubiquitin to target proteins, has been best studied in the context of protein degradation. For example, the ubiquitination-mediated protein turnover has been shown to play an essential role in driving the cell cycle [67]. However, recent research has also revealed a role for ubiquitination in multiple protein-degradation-independent intracellular signaling pathways.
As an example, a protocol for the detection of linear polyubiquitin chains by an anti-linear ubiquitin antibody used by Sasaki et al in their studies can be found here [68]. Advantages and disadvantages of the commonly used immunoblotting-based methods, as well as the suggestion for their optimization, are highlighted in a recent review [69]. The review includes recommendations on sample preparation, separation, detection and identification of ubiquitin chains, as well as on the analysis of the ubiquitin chain topology.
Alternatively, ubiquitinated proteins can be detected by affinity purification. Two such approaches are becoming more popular: ligase trapping [70], and the TUBEs (Tandem Ubiquitin Binding Entity) technique [71]. In ligase trapping, ubiquitin ligases are fused to a polyubiquitin-binding domain, which allows the isolation of specific ubiquitinated substrates that can be further analyzed by mass spectrometry or western blotting. Similarly, TUBEs, which are engineered tagged sequences of four ubiquitin-binding domains linked together via flexible likers, are used to affinity purify polyubiquitinated proteins from cell extracts [71, 72],]. The TUBEs protect the ubiquitinated protein from degradation and de-ubiquitination, thus facilitating the identification of ubiquitination target sites as, for example, specific substrates of ubiquitin ligases [73]. TUBEs for protein purification (agarose-TUBEs, GST- and His6-TUBEs), immunohistochemistry (fluorescent TUBEs), or Far Western (ligand) blotting (biotin-TUBEs) are now available for researchers from LifeSciences.
The diversity of ubiquitin-mediated signaling depends on the multiple ways in which a target protein can be modified by the ubiquitin – monoubiquitination at one or several sites, and polyubiquitination via several possible linkages [74]. Substrate proteins are linked to ubiquitin via seven distinct ubiquitin lysine residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48 and Lys63). Formation of a polyubiquitin chain happens when a lysine residue of ubiquitin is linked to the carboxy-terminal glycine of another ubiquitin. Thus the functional outcome of polyubiquitination depends on the lysine residue within ubiquitin that is used. This is, in turn, determined by the specific E2 (ubiquitin-conjugating enzyme) or E3 (ubiquitin ligase), but not the E1 (ubiquitin-activating enzyme) (Fig 4A). The generation of polyubiquitin chains with specific lysine linkages creates a unique binding surface for the specialized ubiquitin-binding domains of interacting proteins.
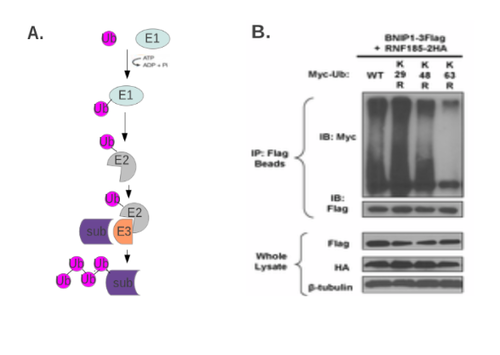
Methods for assaying ubiquitination can reveal the ubiquitination at specific lysine residues or confirm the role of specific E3s (or E2s) in the modification of the target protein. The most straight-forward and commonly accepted way to assess ubiquitination in vivo is to immunoprecipitate the putatively ubiquitinated protein, and subject it to SDS-PAGE/Western blotting to observe the laddering typical of ubiquitinated proteins. The target protein, E3 ligase, and even ubiquitin are often ectopically expressed to determine the effects of mutation (Fig 4B). Inhibitors of proteasomal degradation, such as MG132, are frequently used to analyze the in vivo K48 (degradation-targeting) ubiquitination of target proteins.
In vitro ubiquitination assays (where the target protein, ubiquitin, and the E1, E2, and E3 are provided) can be used, in conjunction with SDS-PAGE followed by Western blotting to more directly assay the ubiquitination of the protein in question. Mutations of lysine residues within the protein in question can serve to determine the site of ubiquitin attachment. Conversely, in the case of poly-ubiquitination, utilizing ubiquitin with mutations of specific lysine residues can provide information about the specific ubiquitin linkage. To discover novel ubiquitinated proteins, ubiquitin-mediated protein purification followed by mass spectrometric analysis can be performed [75].
Epigenetic modifications (such as phosphorylation, ubiquitination, acetylation, and methylation) of the conserved core histones H2A, H2B, H3, and H4 play an important regulatory role in gene expression [76]. For example, acetylation and methylation of lysine residues on the N-terminal histone tails mediate the formation of chromatin domains, such as euchromatin and heterochromatin, thus directly mediating gene silencing. Assaying the modification status of core histones has been an important technique in analyzing the regulation of gene expression [77]. Detailed histone modification research methods are discussed here.
Acetylation and methylation of other proteins have also been studied, for example, K49 acetylation of beta-catenin [78], or α-tubulin acetylation [79]. M Oginuma et al detected the acetylated beta-catenin in Western blots using a specific antibody ( 9534 from Cell Signaling Technology) [78].
Extensive collections of antibodies are commercially available for all known histone modification sites. Using these antibodies, a chromatin immunoprecipitation (ChIP) experiment can be performed [80]. Briefly, the cell or tissue sample is treated with a cross-linking agent such as formaldehyde to cross-link histones to chromatin. The chromatin is sonicated or treated with a nuclease to generate short DNA fragments. An immunoprecipitation (IP) step is then performed using an antibody against the desired histone modification. If the putative histone modification site within a gene or promoter is known, PCR can be used to quantify the presence of the modification in question at a given site. (Fig 5A). Alternatively, this method can be used to identify the histone modification site within the general promoter region, as well as the kinetics of histone modification at that site (Fig 5B).
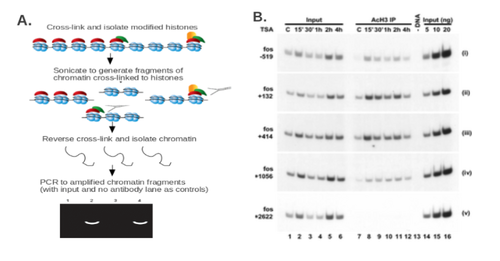
If the site of histone modification is not known, a ChIP-on-chip can be performed to identify the regions in the genome carrying specific histone modifications [81]. In this setup, the DNA fragments in the IP input and the IP itself are labeled with different fluorophores. The samples are then passed over a chip with DNA probes, and the enrichment of a given DNA fragment in the IP fraction correlates with the localization of the modified histone in question to a given site in the genome.
ChIP and ChIP-on-chip experiments can provide information about histone modification at a promoter of interest, the kinetics of change in the modification status, as well as the location of histone modifications with respect to a gene or regulatory sequence, or throughout the genome. These epigenetic changes can shed light both on gene regulation in normal function and the dysregulation that occurs in disease.
Glycosylation, or the attachment of one of a large number of glycan groups, is predicted to occur in close to 50% of all proteins and plays a role in biological processes as diverse as embryonic development, cell division, and regulation of protein structure. The glycosylation status of a wide array of proteins changes significantly during normal physiological events, and in diseases (for example inflammation, sepsis, and cancer). Thus, assays for protein glycosylation can be informative in research aimed both at disease prognosis and therapeutic purposes.
The two main types of protein glycosylation are N-glycosylation (in which the glycan is attached to an asparagine) and O-glycosylation (in which the glycan is attached to a serine or threonine). Glycosylation differs from protein modifications discussed above in that a vast array of modifying glycans has been described. Analysis of protein glycosylation is aimed at identifying the glycan group, the modified protein, or the site of attachment. In general, glycosylation analysis can be performed using three different approaches, as outlined in Fig 6A: the analysis of chemically or enzymatically released glycans themselves, the characterization of glycopeptides following a tryptic digest, or the characterization of glycans in intact glycoproteins.
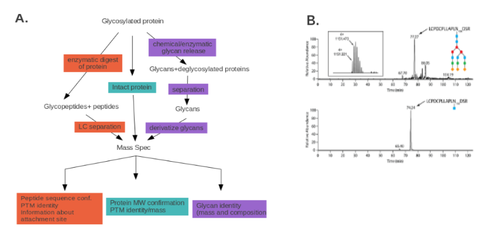
For the analysis of glycans, mass spectrometry analysis, sometimes coupled with an HPLC step, can be utilized. The sugar groups are first released from the glycoprotein either enzymatically (peptide N glycosidase A or F) or chemically (hydrazinolysis). LC/MS analysis can then be performed directly on the released oligosaccharides (for example, [82] ). Alternatively, the reducing terminus produced by enzymatic or chemical cleavage can be tagged with a fluorophore label, and the fluorophore-labeled glycans can be analyzed by a number of possible HPLC methods, such as hydrophilic interaction HPLC [83].
An added fluorophore or chromophore at the glycan’s reducing end allows for their separation by electrophoresis or chromatography, and for their quantitation. Currently, solid-phase based approaches such as glycoprotein immobilization or on-tissue glycan mass spectrometry imaging are used alongside the more common solution-based analyses. A recent review of the review, methods for glycan isolation, identification, and quantitation can be found here [84].
Reagents with high affinity for glycans, like lectins and glycan-binding antibodies, have been used mostly to detect glycosylated proteins in clinical samples. Recent advances involve the use of glycan arrays, lectin engineering, and quantitative lectin measurements. Some of the challenges and advantages of these new tools for biological samples analysis have been reviewed recently [85].
While the methods described above provide information on the specific glycans, they are not informative when it comes to the identity of the protein carrying a specific modification or the site of attachment. To obtain this information, endoproteinase cleavage is performed (e.g., a tryptic digest), and the glycopeptides are analyzed by LC/MS or HILIC-MS/MS. This method can provide information on either all proteins carrying a specific glycan moiety or the site within a given protein where the glycan is attached [86]. For example, in Fig 6B, the effect of incubation with Cc5 bacteria on the glycosylation status of specific fetuin residues is assayed, following the tryptic digest, by LC-MS.
For some glycopeptides, subjecting the intact protein to mass spectrometric analysis can provide data about the protein identity and its glycosylation status. This method, however, is dependent on the biochemical properties of the glycopeptide and often provides a lower level of resolution than the two methods described above.
Multiple reviews of the major developments in the field of glycoproteomics that focus on the most recent advancements (2012-2015) that involve the use of MS-based approaches have been published [87-90].

Sumoylation, the addition of SUMOs (small ubiquitin-like modifiers) to proteins, is a post-translational modification similar to ubiquitination. As all other PTMs, sumoylation can affect a protein’s structure, subcellular localization, and function. As for the identification of other PTMs, mass-spectrometry is a useful tool despite its limitations. As of June 2016, a few recent reviews of the MS-based techniques for PTMs analysis have focused not only on the more studied types (phosphorylation, glycosylation, acetylation, methylation or ubiquitination) but also on the less common modifications, like sumoylation [41, 91].
Several other methods have been proposed. Among them, the use of SUMO-specific molecular traps, called SUBEs (SUMO Binding Entities), that is similar to the previously described TUBEs (see Ubiquitination above) and for which a methodological overview has been published [72] was important in the understanding of tumor-suppressing pathways in human cells [92].
Additionally, the Protease-Reliant Identification of SUMO Modification (PRISM) method, which allows for detection of SUMOylated proteins and the identification of SUMOylation sites, has been used [93]. The method involves the chemical blocking of all free lysines, with SUMO-specific proteases cleavage, and MS-based identification of the unblocked lysines (Figure 7).
- Hazzalin C, Mahadevan L. Dynamic acetylation of all lysine 4-methylated histone H3 in the mouse nucleus: analysis at c-fos and c-jun. PLoS Biol. 2005;3:e393 pubmed
- Controlled vocabulary of posttranslational modifications. Available from: www.uniprot.org/help/controlled_vocabulary
- RESID Database at PIR. Available from: pir.georgetown.edu/resid/
- Mann M, Hendrickson R, Pandey A. Analysis of proteins and proteomes by mass spectrometry. Annu Rev Biochem. 2001;70:437-73 pubmed
- Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312:212-7 pubmed
- Korfmacher W. Principles and applications of LC-MS in new drug discovery. Drug Discov Today. 2005;10:1357-67 pubmed
- Verbeck G, Ruotolo B, Sawyer H, Gillig K, Russell D. A fundamental introduction to ion mobility mass spectrometry applied to the analysis of biomolecules. J Biomol Tech. 2002;13:56-61 pubmed
- Lim Y. Mining the tumor phosphoproteome for cancer markers. Clin Cancer Res. 2005;11:3163-9 pubmed
- Cohen P. The regulation of protein function by multisite phosphorylation--a 25 year update. Trends Biochem Sci. 2000;25:596-601 pubmed
- Hastie C, McLauchlan H, Cohen P. Assay of protein kinases using radiolabeled ATP: a protocol. Nat Protoc. 2006;1:968-71 pubmed
- Peck S. Analysis of protein phosphorylation: methods and strategies for studying kinases and substrates. Plant J. 2006;45:512-22 pubmed
- Zhang H, Zha X, Tan Y, Hornbeck P, Mastrangelo A, Alessi D, et al. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J Biol Chem. 2002;277:39379-87 pubmed
- Blaydes J, Vojtesek B, Bloomberg G, Hupp T. The development and use of phospho-specific antibodies to study protein phosphorylation. Methods Mol Biol. 2000;99:177-89 pubmed
- Takeda H, Kawasaki A, Takahashi M, Yamada A, Koike T. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry of phosphorylated compounds using a novel phosphate capture molecule. Rapid Commun Mass Spectrom. 2003;17:2075-81 pubmed
- Tyers M, Jorgensen P. Proteolysis and the cell cycle: with this RING I do thee destroy. Curr Opin Genet Dev. 2000;10:54-64 pubmed
- Tomlinson E, Palaniyappan N, Tooth D, Layfield R. Methods for the purification of ubiquitinated proteins. Proteomics. 2007;7:1016-22 pubmed
- Grant P. A tale of histone modifications. Genome Biol. 2001;2:REVIEWS0003 pubmed
- Spencer V, Sun J, Li L, Davie J. Chromatin immunoprecipitation: a tool for studying histone acetylation and transcription factor binding. Methods. 2003;31:67-75 pubmed
- Buck M, Lieb J. ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics. 2004;83:349-60 pubmed
- Wuhrer M, Deelder A, Hokke C. Protein glycosylation analysis by liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;825:124-33 pubmed
- Materials and Methods [ISSN : 2329-5139] is a unique online journal with regularly updated review articles on laboratory materials and methods. If you are interested in contributing a manuscript or suggesting a topic, please leave us feedback.
- method
- Aminoacyl-tRNA Synthetases
- Extracellular Traps – NETosis and METosis
- Histone Modification
- Identification of Ubiquitinated Proteins
- Incorporating Unnatural Amino Acids into Recombinant Proteins in Living Cells
- Phosphotyrosine Antibody
- Protein Companies
- Quantitative Bioanalysis of Proteins by Mass Spectrometry