Insoluble aggregates of proteins were initially not considered as worthwhile objects of research. They were difficult to investigate and did not appear to be related to processes of life. When amyloids, a special set of such aggregates, was found deposited in cells and tissues and to be involved in diseases, strong interest has arisen to characterize such objects. The study of the aggregates has remained difficult for a long time, however, particularly as long as only a few methods were available.
By now, many analysis methods have been established, and it is important that the knowledge of old method is maintained. Because only through thoughtful combinations of different methods more insight into subunits, structure, or formation and function of aggregated species can be gained. Here, the methods for solubilization in buffer with pH-changes and various denaturants and the application of analytical techniques like ultracentrifugation, size-exclusion chromatography, field flow fractionation, SDS-PAGE, light scattering, mass spectrometry, chemical crosslinking, and electron microscopy are listed and described. Additionally, immunological methods specific for aggregated proteins are presented as well as analysis techniques for the toxicity of amyloids. Seeding experiments and methods to assess agglomeration by measurement of turbidity and theoretical methods of molecular dynamics simulation and of fluctuating thermodynamics calculations are included.
Valuable insights in evolution, function and structure of protein aggregates has been established by combining the findings from various methods analysing the components of aggregates, the surface and structure of the aggregated species. As a result, the understanding of varying size of protein aggregates from oligomers to highly multimeric species was found. By high-resolution methods it was achieved to specify a typical amyloid structure, the cross-β-fold. In the future, more insights into the functions of protein aggregates occurring in vivo in many different organisms, might be gained.
Proteins are linear strings formed by chains of amino acids that can fold into one or more specific three-dimensional forms. They can have different length up to over 30,000 amino acids. Some of them adopt folds which are stable for a lifetime like a stone, while others are more flexible allowing refolding and rearrangement. Proteins are diversified molecules in life forming both, the machineries and the major structural components of cells and bodies. Many are soluble constituents of the cytosole, others are integrated in membranes, and some are deposited in cells and tissue as insoluble aggregates. The analysis of such protein aggregates is still often remaining a challenge although much progress has been made during the last 100 years since their discovery benefitting from the development of new biochemical techniques.

By definition, an aggregate is a whole formed by combining several separate elements. A molecular protein aggregate can be defined as cohesion of many distinct single protein units. It is an assembly formed from a single type of protein monomers or a mixture of different types. Often it is thought that aggregates are only found as protein clumbs that cannot be solubilized within a certain solvent or buffer. However, all multimeric proteins consisting of more than one polypeptide chain and therefore have a quaternary protein structure, where protein subunits interact are a kind of aggregate. Accordingly, protein machineries consisting of several different subunits are protein aggregates, like for example the ribosome, polymerases, protein channels or the proteasome.

In many neurodegenerative diseases, the so-called amyloidosis, it was observed that single misfolded proteins are assembling into water-insoluble large protein aggregates. These misfolded, amyloidogenic proteins are thought to be the causative agent of the diseases [1]. In Alzheimer disease and in prion disease, the aggregates of tau and prion proteins are forming so-called amyloid plaques that occur in the brain. Other proteins, such as α-synuclein, apolipoproteins, insulin, lysozyme and lactoferrin can also form such highly ordered amyloids [2], see Figure 1. TDP-43 aggregation, occuring in amyotrophic lateral sclerosis, frontotemporal dementia, Alzheimer’s disease, and limbic predominant age-related TDP-43 encephalopathy, goes through a process of liquid-liquid phase separation and anisosomes [3]. In addition, CCT2, a chaperonin subunit, has been identified to regulate the clearance of aggregation-prone proteins [4].

Initially characterised as an aberrant protein fold associated with mammalian disease, in the last two decades, functional amyloids have been described in almost all biological systems, from viruses, to bacteria and archaea, to humans [5]. Protein aggregation is causally linked to protein folding and stability [6]. It occurs in vivo and in vitro. In the biochemical literature, it is often stated that such protein aggregation is the process by which misfolded proteins adopt a conformation favouring polymerization to organized fibrils [7].

Besides the amyloids another in vivo form of biological of protein aggregations are the intracellular inclusion bodies. They are dense protein agglomerates that often occur during overexpression of proteins in bacteria. Although they have an amorphous appearance high-resolution studies disclosed amyloid-type structures formed by short aggregation initiation segments [8].
By now, the aggregation of intrinsically disordered proteins or misfolded proteins and their intra- or extracellular accumulation is a recognized biological phenomenon [9]. It is not known yet, however, if this clustering or clumping of proteins serves a definite function. Already, in 1931 it had been postulated that nature may exploit the capability of proteins to coagulate (Anson and Mirsky’s, 1931). In line with this view proposals of roles for amyloid structures such as protection, adhesion, and storage have emerged [8]. Similarly, the collection of proteins and their sequestration in aggresomes, special inclusion bodies, has been seen as a potential benefit for the cell [10]. The compact β-sheet-structure (Figure 2) seen in amyloids is also found in other aggregated protein providing a structural entity found in disease and health [11].

In all forms of life many proteins are naturally assembled to aggregated structures. Examples are the proteins of the shells of viruses, as for example a Dengue virus (Figure 3). Also, the muscle proteins actin and myosin are arranged in ordered filaments. Collagen is a triple helix of three made up of individual helices (Figure 4). The clustering of single protein chains often gives rise to new functions. So, elastin is formed as helical structure crosslinked by desmosine, four crosslinked lysine amino acids, and subsequently can be stretched and relaxed [12]. Elastin-like peptides are derivatives of tropoelastin with a unique property that allows them to stay soluble below a certain critical temperature but reversibly form aggregates above that temperature.
Other examples for protein aggregates are microtubules, the polymers of α- and β-tubulin that are major components of the cytoskeleton of eukaryotic cells (Figure 5) [13]. They are forming rigid hollow rods which change their length by ordered bonding and releasing of protein units, thereby maintaining, and altering the cellular architecture. This is an example of an ordered protein aggregate where the increase and decrease of the size of the aggregated amount of protein is causally related to its function.
An extracellular protein prone to undergo intermolecular aggregation is fibrin. Generated from its precursor in the blood during the coagulation cascade it produces by intermolecular crosslinking a multimeric insoluble mesh to hinder the loss of blood [14].
A further very systematic structured form of protein aggregates are protein crystals. There the subunits are ordered in regular arrays. The individual protein molecules are packed systematically in porous arrays [15]. Mostly protein crystallization is done experimentally in vitro, and the formed crystals are used for high resolution of molecular structures by X-ray diffraction methods. When the subunits in the crystal are sufficiently ordered, good diffraction for structural analysis is obtained.

Some proteins naturally form crystalline arrays in vivo, like aquaporin in the lens of the eye [16]. Others, the β-crystallins that are chaperones, transform into high molecular weight light-scattering aggregates with increasing age what results in cataracts [17].

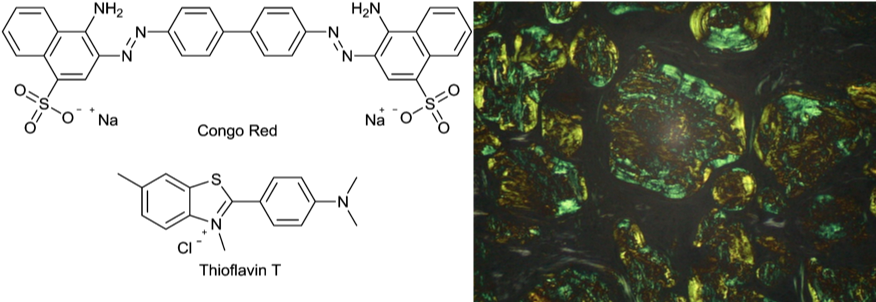
Amyloid was first mentioned over 160 years ago to describe white densities of protein aggregate in autopsied organs of patients who had died of chronic diseases [18]. At that time there was the belief that these clumps represented deposits of starch, therefore the name amyloid from the latin word amylum for starch. Now it is known that these deposits consist of protein aggregates, dense accumulations that have special staining characteristics. This dye binding ability is a diagnostic feature of amyloid deposits. In tissue amyloid reacts with Congo red and shows a characteristic green birefringence in polarized light [19] (Figure 6). Similarly, the fluorescent dye Thioflavin-T identifies amyloid fibrils both in vivo and in vitro and is used as standard and as a powerful tool for selective staining by high fluorescence emission [20, 21].

With time the term amyloid has acquired a classifying meaning beyond its original histopathological association and is also no longer always associated with disease [22]. It now stands for a class of nanofibers which can be formed by most proteins upon their adoption of an unfolded structure and subsequent polymerization via intermolecular β-sheet formation. It has been found that unstructured protein agglomerates, so-called amorphous protein aggregates were found to contain partly also amyloid-like structures [11] (Figure 7). More and more it is apparent that the ability to form amyloid structures is not an unusual feature of the small number of proteins associated with diseases but is instead a general property of polypeptide chains [23].
Similar structures are now found for a lot of amyloid fibrils. However, there is no universal amyloid fold [24]. There is the so-called cross-β structure as a general feature (Figure 2), whereby many amyloid fibrils have a parallel β-sheet structure, and some have antiparallel β-sheets. Further, more subtle structural differences among amyloids exist and are found as more and more structures are solved. Additionally, conformational plasticity was found for amyloids reflecting their ability to adopt more than one stable tertiary fold.
During aggregation of proteins new chemical bonds are formed. The intermolecular connections in aggregated proteins are in fact of the same type as the non-covalent intramolecular bonds in single proteins molecules (Scheraga in “The proteins I, p. 479). Hydrogen bonds occur between peptide groups and side chain groups, that can form between neutral polar groups like OH-groups and =O groups as well as between charged groups. Hydrophobic bonds are found between two non-polar groups, that can be side chains or backbone groups. All these single bonds are individual weak bonds, but by the formation of many bonds cooperative interaction is seen. To be as strong as one covalent bond, about one hundred hydrophobic or Van der Waals bonds are needed. The strength of ionic bonds and hydrogen bonds lies in between covalent and hydrophobic ones.
The bonds are dependent on buffer and pH, ionic strength, temperature, and solvent composition. Aggregated proteins can be lower in free energy than the soluble protein monomers. For example, upon association free energy can arise from formation of hydrogen bonds between side chain groups that associate as has been observed during the formation of polymer from fibrin monomers [25].
| Protein solubility | Changing buffer conditions Salt precipitation Denaturation Solubilization with detergent and lipid |
| Aggregate size | Ultracentrifugation Size-exclusion chromatography Field flow fractionation Light scattering |
| Aggregate components | Sodium-dodecylsulfate-polyacrylamide gel electrophoresis Mass spectrometry |
| Subunit interaction | Chemical crosslinking |
| Aggregate surface and structure | Light and fluorescence microscopy of stained samples X-ray crystallography X-ray fiber diffraction Electron microscopy Atomic force microscopy Solid-state NMR-spectroscopy Circular dichroism Immunological techniques Protease sensitivity |
| Aggregate formation | Seeding experiments Turbidity Molecular dynamic simulations Fluctuating thermodynamics analysis |
| Aggregate toxicity | Cell-based assays Infectivity assays |
Here, several techniques that can be used to analyse protein aggregates will be enumerated and described (Table 1). There is not one analytical method or approach that can answer all situations of aggregation. Therefore, here the different methods useful for the analysis of protein aggregates are outlined.
Generally, it will depend on the question to be answered, if one wants to know the subunits of an protein clump, the size or to get information on the structure, or maybe especially on the binding of the protein components to each other another analysis method is necessary. A question might be how the formation of amyloids could be monitored or one is looking for a mechanism how an aggregate can restructure other protein monomers to stick to it.
Often a method comes up with the tools, the machines, and the technical equipment. Some methods only work for a defined size range of aggregates, others are specialized on surface or to analyse the subunits. For each of the methods it will only shortly be described how they work, especially in case for protein aggregates. More information can be found in special articles on each method alone. There is a difference if an unknown sample should be examined or a pre-analysed amyloids or viruses investigated further, for example.
Each method is like having a special eyeglass and one will see special details as each technique is different in what can be detected and what are the limits. Sometimes knowledge on the sample is necessary before starting with the method.
The list of methods here to analyse aggregated proteins is not complete as always variations and new techniques arise. However, here a broad overview is given.
In former times and still now a first step of analysis is often fractionation of proteins depending on solubility. Changing individually one of the following parameters pH, ionic strength or temperature gives data on the conditions when a single protein stays soluble or when a protein aggregate is solubilized. There exists also a systematic approach to select a buffer, in which the type of chemical which best improves solubility of proteins is first determined, followed by identifying the optimal chemical and its most effective concentration [26]. Chemical additives can additionally be assessed.
Also, different solvents like chloroform or methanol have proofed useful for the solubilization of certain proteins. A lot has been done with solvents in the early days of protein investigation and the results can be found in the first edition from 1954 of “The proteins” by the editor Hans Neurath.
Generally, native folded proteins aggregate when they are salted out for example with ammonium chloride what leads to isoelectric precipitation when the net charge is zero [6]. A high amount of the salt is just added to the protein in solution. The point of precipitation depends always also on the protein concentration. In salting out precipitates the native conformation of a protein can be retained [6].
Mostly amorphous aggregates can be dissolved by buffer change in contrast to the pathological amyloid aggregates that dissociate only in the presence of high concentration of denaturants like urea or guanidine hydrochloride or detergents like SDS (sodium dodecyl sulfate). For inclusion bodies it is known that they dissolve in urea or guanidine hydrochloride [27]. The denaturants disrupt many of the weak intra- or intermolecular bonds of protein molecules like hydrogen bonds and form new bonds with the small denaturant molecules. The denatured proteins have a more random, loose structure.
In contrast the detergent SDS has another mechanism of unfolding proteins that is still not well known. Molecular dynamic simulations showed for two proteins that they fast unfold when boiled with SDS. In the final state of unfolding, the proteins are found to wrap around SDS micelles. As the proteins then form long strings, the SDS bound to the protein is correlated to the size of the protein and therefore upon loading on a polyacrylamide gel proteins are separated approximately to their molecular weight.
For aggregated prion protein in form of insoluble amyloid rods that cannot be solubilized by nondenaturing detergents, it has been shown, that it was rendered soluble by a mixture of detergent and lipid in fact cholate and phospholipids were used [28]. Upon this treatment so called detergent-lipid complexes were formed that stayed in the soluble fraction upon ultracentrifugation at 100’000 g. Removal of the cholate resulted in the formation of closed liposomes.
An obstacle for measuring the size or the number of subunits of a protein aggregate is, that a protein clump is not a homogenous species. Often single proteins assemble to dimers and other oligomers that than form larger aggregates of different size.
Differential centrifugation is used to separate whole cell homogenates into single components (Figure 8). Therefore, repeated centrifugation at progressively higher speed is performed. The fractionation happens based on size and density. The largest and densest components experience the greatest centrifugal force and sediment most rapidly. Protein aggregates are usually pelleted by very high-speed centrifugation, so-called ultracentrifugation.
Ultracentrifuges can be used either to measure the velocity of movement of macromolecules in a solution under the influence of a centrifugal field or to determine the distribution of the macromolecules in a centrifuge cell which is rotating at comparatively low speeds. In the latter after a fixed period, an equilibrium state is established in which the concentration of solute at each level in the cell no longer varies. Ultracentrifugation is used to separate protein monomers from aggregate forms under a broad range of solution conditions [29]. Careful application of experiments with ultracentrifuges is obligatory as the use of the instrumentation is difficult and the data analysis process complex. In the analytical ultracentrifuge protein concentration is measured at specific radial positions by a UV absorption or an interference measuring optical system and at specific time points during the sedimentation process. Here precision and accuracy are indispensable for exact detection and subsequent data analysis, especially if trace amounts of soluble aggregates are measured.
Density gradients are used to separate biological material according to mass. Loading of cell fraction from bacteria overexpressing a protein on a sucrose gradient can yield sub-classes of aggregates [30]. Lower and larger mass aggregates can be seen and after recovery these fractions can be further analysed biophysically, by electron microscopy of the macro-structure, for disaggregation by molecular chaperones. Often experiments with analytical ultracentrifugation are combined with size-exclusion chromatography and field flow fractionation to provide complementary information.
Size exclusion chromatography is one of the most popular analytical methods widely employed for the characterization of proteins, capable of detecting lower molecular weight oligomers and a powerful technique for the qualitative and quantitative evaluation of aggregates [31, 32]. Size exclusion chromatography is performed with columns consisting of spherical porous particles with a carefully controlled pore size. This is the stationary phase. The proteins flow with the mobile phase, an aqueous buffer dependent on their molecular size through the pores, if the molecule is small enough, or around the spherical particles. This leads to a separation of molecules according to their hydrodynamic radius that is correlated with the size of the proteins. The larger a molecule, the sooner it elutes and the smaller a molecule the later it elutes. Different polymeric resins, such as agar, agarose, polyacrylamide and many others exist [33]. UV is the predominant mode of detection, that can be adjusted to different wavelengths. Also, fluorescence detectors and multi-angle light scattering detectors or combinations thereof are in use.
For better understanding size exclusion chromatography can be combined with online detection by native electrospray ionization mass spectrometry [32]. This combination allows a distinction between the oligomers present in solution and those formed during the experiment that are artifacts of mass spectrometry. Size exclusion chromatography is used as quality control for the detection of aggregates in protein-based drugs that are critical due to their potential immunogenicity [34, 35].
Particularly for monitoring the large and insoluble aggregates field flow fractionation is a very useful method [29]. It separates macromolecules based on their differences of diffusion coefficients [36]. An external field that is perpendicular to the flow channel controls the retention and separation of the samples. Flow, sedimentation, electrical, and thermal field fractionation use different fields, whereby flow technology has been widely used and is most suitable for separation of protein aggregates.
For field flow fractionation experiments samples are injected into a thin, elongated flow chamber, the channel. A semipermeable membrane that is permeable for the elution buffer but not for the samples, is used as accumulation wall to prevent samples from exiting the channel. In a first step the samples are focused into a narrow band by two opposite flows, and then in a second step eluted with a continuation of flow into a detector. Smaller molecules have larger diffusion coefficients and tend to diffuse and concentrate closer to the centre of laminar flow where the flow rate along the length of channel is faster. This results in an earlier elution of smaller molecules compared to larger ones or aggregates, as these will be closer to the membrane owing to the relatively small values of their diffusion coefficient.
The sample range to be analysed by field flow fractionation extends from about 1 nanometer to more than 100 micrometers and incorporates both simple and complex macromaterials [36]. Additionally, to size and mass, properties like density, charge, diffusivity, and thickness of adsorbed layers can be determined.
When a protein sample contains visible particulates, dynamic light scattering is a highly effective analysis method [37]. This technique is routinely used in biology laboratories to detect aggregates in macromolecular solutions, to determine the size of proteins and other complexes [38]. A dissolved or suspended sample is irradiated by a laser and the amount of scattered light from the laser is determined and analyzed according to temporal fluctuations and particle size is calculated by autocorrelation functions. It is important to keep variables like temperature, solvent viscosity, and inter-particle interactions constant as these may also influence particle size determination. Further, another variation of the method, differential static light scattering, can be used for the assessment of protein aggregates [39].
Additionally, dynamic light scattering has been used to detect detergent-mediated non-specific protein aggregation [40], as well as formation of protein aggregation by heat and suppression of aggregation by α-crystallin, one of the small heat shock proteins [41].
The simplest method to determine subunits and their molecular weight is polyacrylamide gel electrophoresis (PAGE), mostly used is SDS-PAGE with the denaturant sodium dodecyl sulfate. It is well-known since long time, markers are available. In this method protein aggregates are disassembled during heat treatment with SDS in reducing conditions. Not only quarternary structures where different subunits hold together are disassembled but also secondary and tertiary structures of proteins are denatured. On the gel bands for different sized molecules can be detected. Sometimes dimers and other oligomers are heat-stable and can be detected on the gel.
Native PAGE without SDS is more complicated and does not give direct molecular weight information because of protein conformation or when an aggregate is too large to enter the gel. Often SDS-PAGE is followed by Western blotting if the protein is known and antibodies are available.
Mass spectrometry is an analytical technique that measures the mass-to-charge ratio of ions. It has been widely used for proteins [42]. During a typical experiment a sample, which can be solid, liquid, or gaseous, is ionized, often by bombarding it with electrons. The molecules in the sample are charged and might additionally break into charged fragments. These ions are then separated according to their mass-to-charge ratio when they are accelerated in an electric or magnetic field. If ions have the same mass-to-charge ratio they will have the same amount of deflection. This method is important for the accurate mass determination and characterization of proteins. To study insoluble proteins and to analyse structural properties that occur in aggregated samples mass spectrometry can be used on amyloid fibrils for example after hydrogen/deuterium exchange [43]. Also screening for inhibitors of the aggregation process has been performed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry.
Native mass spectrometry is an emerging method to identify and characterize protein complexes, macromolecular species and small oligomeric protein aggregates [44]. Electrophoretic and chromatographic methods can be coupled to mass spectrometry to obtain fast combined data.
Crosslinking of single amino acid side chains in the aggregated protein followed by analysis of where the links have been inserted, can give direct information on amino acids which are close in the aggregated structure.
Crosslinking of proteins can be done by treatment with formaldehyde. The chemical introduces CH2-bridges between closely lying lysine side chains with other amino acid groups like asparagine, glutamine, arginine, phenylalanine, tryptophan, or histidine in a two-step mechanism. High amounts of formaldehyde can lead to aggregates that are so much crosslinked that they cannot be solubilized any more, whereas the use of limited amounts can lead to soluble components where the structure is stabilized against denaturants.
Additionally, chemical crosslinking has already been performed on amyloid Aβ by photo-induced crosslinking of unmodified proteins with tris-bipyridylRu(II) activated by UV-light [45, 46]. More on the method of chemical crosslinking of proteins can be found in “Advances in Experimental Medicine and Biology”, Volume 86B, Protein Crosslinking, 1976.
Crosslinking can be used as an analysis method, but it is also used in nature to introduce new features to proteins. By this method the introduction of distinct bonds at specific places creates a new entity that has different properties as a result of the union. In the case of proteins, such crosslinking often results in important changes in chemical, physical, functional, nutritional, and biomedical properties, besides physical properties simply related to molecular size and shape.
The interaction of protein subunits is also analysed by structural methods where with high resolution techniques atomic interactions are investigated. These methods are described in the following part.
To detect protein aggregates specifically under a microscope often staining with specific dyes is performed to make them clearly visible. Well-known are Congo red and Thioflavin T. They have been used since many years to investigate amyloid formation.
Congo red is an organic compound, an azo dye (Figure 6). Staining with it is a qualitative method used for the identification of amyloids in vitro and in tissue sections [47]. Upon binding an orange-red appearance under a light microscope and the typical apple-green birefringence of Congo red under polarised light stained preparations is indicative of the presence of amyloid fibrils [48]. Congo Red has been used for in vitro investigations and in vivo histological studies using tissue sections. However, there are limitations and the technique must be used carefully or in combination with others.
Thioflavins T is a popular fluorescent dye. Fluorescence is the emission of light by a substance that has absorbed light. Generally, the wavelength of the emitted light is longer and thereby has less energy than the adsorbed wavelength. When using fluorescence microscopy an image is visualized by fluorescence from the analysed material under a microscope.
Fluorescence microscopy of protein aggregates describes a technique, which consists of analysing probes by fluorescence microscopy after staining with dyes that show fluorescence upon binding to protein aggregates [49].
Upon binding of Thioflavin T to amyloid fibrils it shows a great fluorescence enhancement [20, 21] and is therefore often used in biophysical studies of protein aggregation. It is likely that the amyloid presents a binding site for Thioflavin T that sterically fixes the dye and thereby leads to an enhancement of fluorescence. Understanding of Thioflavins T -fibril interactions at an atomic resolution yields insight into amyloid structures, the processes of fibril formation, and helps designing amyloid diagnostics, inhibitors, and therapeutics.
There exist also novel fluorescence-based probes and procedures that might be applied to experiments on amyloid self-assembly in vitro and in vivo [50-52].
Most macromolecular structures of proteins that have been solved with high resolution so far, have been analysed by X-ray crystallography. A protein crystal can be seen in Figure 9. Crystallization of biological macromolecules is a difficult process performed with purified proteins in vitro. It is influenced by many factors, including pH, temperature, ionic strength in the crystallization solution, and even gravity [53]. Only well-ordered crystals can be examined by X-ray diffraction [54]. The X-rays diffract into many specific directions and when the angles and intensities of these diffracted beams are measured. From the three-dimensional picture of the density of electrons within the crystal, the mean positions of the atoms in the crystal are determined.
As protein crystallization is an established method that started before over 100 years, different special approaches like vapor diffusion, microbatch, microdialysis and free interface diffusion are in use now.
The three-dimensional structures of microcrystals formed from peptide segments of amyloid proteins such as prion protein, insulin and Aβ have been determined by X-ray crystallography [55]. It was indicated that these microcrystals and amyloid fibrils formed by the same peptides have identical structure.
X-ray fibre diffraction is a method where X-rays are used to study the molecular structure of long assemblies of identical protein subunits. The experimental technique and the methods of data analysis lie somewhere between single-crystal diffraction and solution scattering.
As early as in the 1930s structural investigations of amyloids were performed with by X-ray diffraction [56]. Because X-ray fiber diffraction of aligned amyloids yields a characteristic diffraction pattern with a meridional reflection at 4.7 A˚ and an equatorial reflection at 8–11 A˚, the structural organization of the amyloid has been proposed to be the cross β-sheet motif [11, 57].
Electron microscopy yields images of the surface or the inside of an object. In comparison to light microscopy the resolution of electron microscopy is much higher, and it can yield a magnification of more than 10’000-fold. Transmission, scanning and reflection electron microscopy are known methods where always a beam of accelerated electrons is used as source of illumination. Negatively stained electron micrographs show for example amorphous protein aggregates (Figure 7). In difference to that amyloid fibrils are composed of long filaments up to several microns in length, nonbranched and with diameters from 6 to 12 nm [57].
Cryo-electron microscopy is a form of transmission electron microscopy [58]. It is an increasingly popular method to elucidate the structures of macromolecular complexes. The technique involves flash-freezing solutions of proteins. The samples cooled to cryogenic temperatures and embedded in an environment of vitreous water. Subsequent bombarding with electrons produces individual microscope images of molecules. These are then used to reconstruct the three-dimensional shape or structure of single protein molecules or aggregates. Resolutions approaching 1.5 Å are possible.
Atomic force microscopy is a type of scanning probe microscopy with very-high-resolution. It is more than 1000 times better than the optical diffraction limit and lies in the order of fractions of a nanometer [59]. During atomic force microscopy the detector measures the deflection or displacement with respect to the equilibrium position of the cantilever and converts it into an electrical signal. From the intensity of this signal that is proportional to the displacement of the cantilever, an image is calculated while the sample is raster scanned.
A great advantage of atomic force microscopy is that untreated samples can be imaged, even tissue samples can be used directly [60]. Specimen can be imaged under physiological conditions [61]. Atomic force microscopy can even be used to characterise single molecules of protein aggregates [62] and thereby gave insight into the molecular basis of a variety of human pathologies, including neurodegenerative disorders.
Magic angle spinning NMR spectroscopy is a method where insoluble protein material can be measured by nuclear magnetic resonance spectroscopy [63, 64]. During nuclear magnetic resonance experiments with proteins a local magnetic fields around atomic nuclei is observed. The sample is placed in a magnetic field and the NMR signal is produced by excitation of the nuclei sample with radio waves. Thereby nuclear magnetic resonance is detected. The intramolecular magnetic field around an atom in a protein molecule changes the resonance frequency, thus giving access to details of the electronic structure of the molecule and its individual functional groups.
By spinning an aggregated protein sample at a frequency of 1 to 130 kHz at the magic angle with respect to the direction of the magnetic field, the broad lines of the solid become narrower, increasing the resolution for better identification and analysis of the spectrum. The high-resolution structure of a peptide fragment of the amyloidogenic protein transthyretin in its fibrillar form has been solved by this method [63].
Circular dichroism spectroscopy is a versatile technique to monitor protein aggregation [65]. The method involves circularly polarized light often in the far-UV range, and measures the differential absorption of left- and right-handed light. Mostly, far-UV circular dichroism is used to investigate the secondary structure of proteins. The spectra show typical forms with characteristic minima and maxima for different secondary structures and unfolded protein.
The possibility to observe conformational changes during protein aggregation in a simple inexpensive method is a feature of circular dichroism that can measure in situ and in real time [66]. Also, amyloid fibrils can be measured by circular dichroism and show a typical single minimum centered around 218 nm characteristic of extensive β-sheet structures [67]. Real time monitoring of protein aggregation during thermal folding and unfolding transition by circular dichroism spectroscopic experiments can be combined with recording turbidity or light scattering data simultaneously. As this requires no additional time or sample data from different techniques can be directly correlated and protein conformational changes analysed [68].
Thermal scanning circular dichroism is used to examine the stability of therapeutic monoclonal antibodies [65]. It is sensitive for early detection of conformational changes. Further it can help to elucidate mechanisms involved in protein aggregation.
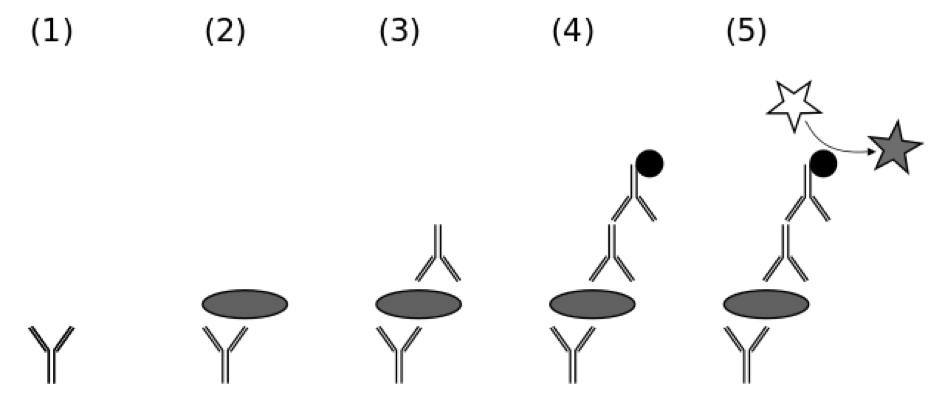
Using antibodies to detect proteins and protein aggregates is particularly useful in several methods. Enzyme-linked immunosorbent assays, ELISAs, are a well-established form of immunoassay, where mostly a first antibody detects the sample, and this is subsequently detected by a second antibody to which a colour reaction is coupled (Figure 10).
Protein aggregate ELISA kits are commercially available. For example, an ELISA to probe human IFN-alpha formulations for the presence of soluble protein aggregates was developed [69]. There exist as well monoclonal anti-prion protein antibodies specific for prion protein dimers of different species or for aggregates of prion protein in the scrapie form and they are used in novel ELISAs [70].
Pull down assays utilize a tag fused to a protein, for example here a tagged antibody. If a protein or an aggregate bind to the antibody it can be captured and “pulled down” when the tag is binding to an affinity resin. This technique helped to understand how protein aggregates are recruited to aggresomes [71]. For immunoprecipitation assays an antibody that specifically binds to a particular protein or aggregate is used. The antibody be coupled to a solid substrate at some point in the procedure. Then with this technique precipitating of a protein out of solution can be performed as is described for immunopurification of pathological prion protein aggregates [72].
Immunofluorescence assays use fluorescent dyes coupled to antibodies.
Proteases have active sites where they catalyze the opening of peptide bonds upon binding of a protein or peptide. Unfolded protein chains can be digested most easily, as they are totally flexible to bind to the protease. Already folded molecules can be more difficult to digest, depending on their size and the sites of digestion as some proteases are sequence specific. A good example where this can be observed is the recombinant form of the prion protein, that consists of a folded C-terminal domain and an unfolded N-terminal tail. When limited amounts of protease are used and the digestion is performed only for a short time, just the N-terminal tail is digested, whereas for chopping of the C-terminal domain longer digestion times or higher amounts of protease are necessary.
When proteins are aggregated, the same features are found. The protease-resistant, scrapie form PrPSc that is aggregated is only digested with high amounts of Proteinase K [73]. To limited amounts of proteinase K it is partially resistant, whereby this resistance depends also on the solute.
Seeding experiments mimic in vitro the aggregation steps occurring during misfolding and aggregation in vivo. They are generally done to imitate amyloid formation. The aggregation process is initiated by adding a small amount of preaggregated species or a chemical like heparin to a mixture with an aggregation-prone protein [74]. Analysis of aggregation to amyloid is often performed by thioflavin T fluorescence. The aggregation leads to a morphology comparable to that seen in vivo in Alzheimers disease and thereby proofed very efficient in seeding the formation of de novo fibrillar structures.
Detection of amyloid growth in solution is commonly carried out by measurement of solution turbidity [22]. This is a low-cost assay procedure based on the intrinsic light scattering properties of the protein aggregate. Basically, turbidity measurement can be done in the UV-range. Most proteins have a peak at about 280 nm because of the absorption of aromatic amino acids that cuts off above at about 320 nm. If you see a broad peak that does not cut off, it is a sign of aggregation. If you can see turbidity already by eye, this is in the visible spectrum, the sample is highly aggregated.
Fitting turbidity data to kinetic models will yield information on aggregate growth and can be compared to growth models with irreversible or reversible nucleation and different kind of fibre associations like end-to-end or lateral. It is also investigated if there is a competition between amyloid formation and amorphous aggregation.
In addition to real experiments simulation have been performed as it is difficult to observe the molecular mechanism of misfolding or aggregate formation directly [75]. Molecular dynamics is a computer simulation method to analyse the physical movement of atoms and molecules. In these experiments given molecules are allowed for a fixed time to interact with dynamic progress of the system and a step-by-step iterative process is applied that forms the basis of the simulation. Combining these results with experimental analyses provides detailed insights into protein folding mechanisms and on the kinetics of the formation of protein aggregates [76].
This approach is a molecular simulation that replaces the conventional thermodynamic characterization of the end states with fluctuating processes that are connected and enables one to analyse variations in the thermodynamic functions that occur during the course of protein conformational changes. The central quantity of this method is the solvent-averaged effective energy, comprising the protein potential energy and the solvation free energy. The entropy of any protein configuration is quantified by the magnitude of fluctuations. Fluctuating thermodynamic analysis proofed useful to explore various forms of aggregate species that are generated through the association of misfolded monomers as well as by soluble oligomers and amyloid fibrils. It has been found that hydration water plays an important role in in protein aggregation.
Mature amyloid fibrils or plaques appear substantially less toxic to cells than the pre-fibrillar aggregates that are their precursors [23]. The toxicity of these early aggregates seems to result from an intrinsic ability to impair fundamental cellular processes by interacting with cellular membranes. It has been stated that the causing of oxidative stress and increase in free Ca2+ might lead to apoptotic or necrotic cell death. For the analysis of these processes special assays have been designed.
Amyloid aggregates or other protein forms can be added to the medium of cultured cells and the effect on the cells be observed directly in a microscope or via binding of fluorescent dyes or other assays [77, 78]. Surprisingly, oligomeric forms of protein seem more toxic than large aggregates. The interaction of the oligomeric protein forms with membranes has been seen in different studies and the biological potential of this species is further investigated.
In animal cells it has been suggested that the aggregation of proteins and the accumulation in inclusion bodies is not diffusion-limited but a process where proteins are specifically delivered to inclusion bodies, so-called aggresomes, by dynein-dependent retrograde transport on microtubules [79].
To protect the cell from unwanted aggregation molecular chaperones and the protein degradation machinery are necessary and appear to be crucial for healthy cells.
Before in vitro assays were available the test for infectivity of aggregated prion protein has been done in vivo in animals like mice or hamsters by inoculation of samples containing aliquots of the aggregated proteins to be tested. In incubation-time assays animals were observed continuously for motor activity and ataxia as early symptoms. From the incubation time infectivity titres could be calculated as well as the mean lethal dose [80, 81].
For aggregated α-synuclein a bimolecular fluorescence complementation assay was developed for use in vivo allowing to determine the abundance of soluble α-synuclein aggregates, most likely oligomers [82]. Insoluble α-synuclein aggregates, most likely fibrils, are detected by filter retardation assay. This is a method that would be ideal for large-scale screening approaches in vivo and might be used to analyze also aggregation of proteins other than α-synuclein. More in vivo assay for protein aggregation can be found in the literature [83, 84].
With the recognition that proteins occur in addition to monomeric and ligated forms as aggregates in vivo and in vitro it is necessary to have the appropriate techniques to investigate these species that are challenging to analyse. Generally, protein aggregation has been seen as abnormal association of proteins that is detected in test tubes as insoluble clumps [85]. However, more and more it is recognized that aggregation occurs under physiological conditions in response to age and stress. It was proposed that proteins that aggregate share several features. When normal physiological conditions change to stress, proteins that are intrinsically aggregation-prone, are affected in a stress-specific manner and start to aggregate.
The Table summarizes all methods described in this essay. Table 1 should give the awareness of what kind of experiments can be done for what kind of question. More methods can naturally be added that might be useful. Here, the techniques are mostly just enumerated and very shortly described, as information is nowadays easily available on the internet. Details can also be found in the references.
For rational understanding of the process of protein aggregation awareness of the organization and architecture of the accumulated proteins is essential. Lately, more and more high-resolution structures of protein aggregates became available. Most important seems to be the cross-β structure of amyloid. This protein fold is characterised by fibrillar morphology, binding of the amyloid-specific dyes Thioflavin T and Congo Red and insolubility [5]. The cross-β sheet entity comprising an indefinitely repeating intermolecular β-sheet motif that is unique among protein folds [86]. It grows by recruitment of the corresponding amyloid protein. Furthermore, the one-dimensional crystal-like repeat in the amyloid provides a structural framework for polymorphisms. By now, the biological importance of microbial amyloid assemblies is recognized by their ubiquity and diverse functionality. Many of the microbial functional amyloids are utilised by pathogens for invasion and maintenance of infection. Understanding the structure and role of amyloids elucidates novel and potentially ancient mechanisms of protein function throughout nature.
Additionally, very surprisingly in bacteria α-helical amyloid-like fibrils were found that are structurally comparable to human β-rich amyloid fibrils [87]. The peptides align perpendicular to the fibril axis in both cases. The α-helical amyloid-like fibrils are formed from the 22-residue phenol-soluble peptide modulin α3 that is secreted by Staphylococcus aureus. The elongated fibrils are highly toxic. Functionally, it was found that in microbe amyloids are often key virulence determinants, however the structural basis for their activity is not yet resolved [87].
The cross-β fold found in amyloid, where β-sheets are formed that staple on each other like corrugated metal, is an extremely dense structure compared with other folds like, where the protein chains align by hydrophobic binding without any water molecules in between. This is likely to be the reason for the insolubility of amyloid aggregates in aqueous buffers. It might also be a reason for its stability and duration through time.
Amyloid fibrils display peculiar membrane-binding behaviour [88]. It was also noted that amyloid assemblies affect the molecular organization of lipid bilayer, that there is competition between fibrillar and monomeric membrane-associating proteins for binding to the lipid surface, and that vice versa lipids have effects on the structural morphology of fibrillar aggregates.
It was discovered that amyloids might have occurred incredibly early in the evolution of life on earth. Very stable protein aggregates could have existed in a prebiotic world and may have been the first functional protein fold in living cells [86]. Scientists were able to show that amyloids can be created amazingly easily from simple amino acids during prebiotic conditions. Alanine, glycine, aspartate and valine can polymerize into peptides and subsequently assemble into ordered amyloid fibers comprising a cross-β-sheet quaternary structure under reaction conditions as they presumably prevailed at the time on the inanimate earth four billion years ago. The discovery confirms the assumption that archetypes of life could have arisen with such amyloids [89]. Current evidence suggests also that the peptide/amyloid world and the RNA world may have originated independently, but at the same time and there are considerable possibilities for interplay between these worlds and for their coevolution.
An aggregated protein in amyloid form seems to be a quite simple form that can self-replicate as for example a prion. However, like viruses they are dependent on living cells to replicate and are therefore not said to be a form of life. Bacteria and organisms with eucaryotic cells that are termed living all consist of one or more formed compartments. These compartments are formed by membranes that give security against the outside world. As the compartmentalization is a fundamental principle of all forms of life, scenarios have been elaborated about the emergence of prebiological compartments on early Earth, in particular about their likely structural characteristics and dynamic features [90]. As amyloids are likely to have emerged at that time point and are prone to interact with membranes they might have played a role in the formation of early compartments and in their replication that consist of a growth and a duplication phase.
In the future, the mechanism how replication, storage and recall of information might be created by these molecules is of great interest. As amyloids were initially discovered in connection with dementia, where abnormalities in recalling information occurs, it will be interesting to see, if the molecular mechanisms have common features in the early systems of life and in our highly developed brains. It is established that long-term memory depends on protein synthesis [91]. For amyloid oligomers of Aβ the inhibition of long-term potentiation was proposed [92]. However, the exact role of protein aggregation in these complicated processes of memory storage is not yet known.
The idea that protein-based beta-sheet molecular structures were the first self-propagating and information-processing biomolecules that evolved already more than three billion years ago was published 2009 [93]. Beside an extraordinary structural robustness, the amyloid fold possesses a unique ability to transmit information by a three-dimensional templating mechanism [94].
When protein is aggregated as amyloid fibril, a large number, over 1000 molecules, are stored in a certain conformation, and there are only at the ends sites for more molecules to attach within this conformation. However, when the protein reacts with lipids and detergent the conformation can be retained in much smaller aggregates, and simultaneously there will be much more, up to 1000 fold more, sites for replication. This has been seen for prion infectivity [28].
The measurement and the control or often the impairment of aggregation continues to be a major concern for biopharmaceutical products [37]. No analytic method will work for all cases of aggregated macromolecules. One has also to think of the fact that the measurement itself may destroy or create aggregates and thereby influence the experiment [37].
In many cases of protein aggregation, the exact atomic-level structural details have yet to be determined, and so far models have been proposed for some core structures [55] that were divided in to three classes: Refolding, Gain-of-Interaction, and Natively Disordered. All these models suggest explanations for some of the common amyloid fibril properties. Gain-of-Interaction models with a cross-β spine fit a wider range of properties than the others.
Protein aggregation in neurodegenerative diseases has been shown to be influenced by post-translational modifications of the aggregating proteins, for example phosphorylation of α-synuclein, huntingtin or tau, isoaspartate formation in Aβ, and acetylation of TAR DNA-binding protein-43 [1]. Still a lot must be learned about the mechanisms by which proteins associate and aggregate and what the meaning in physiology is.
For complete understanding of protein aggregation techniques that can view the process in vivo are especially useful. This still is sometimes difficult, as protein aggregates are not homogenous, often labelling molecules are necessary to observe in vivo processes and they might influence the in vivo action.
Putting together the output of all techniques so far lead to the current knowledge on protein aggregation as a process that is used in vivo and sometimes leads to disease. All further examinations can build up on the understanding that has been gained so far. High-resolution techniques are the basis of understanding the detailed structures and differences in similar but slightly different forms of amyloids.
As we cannot see directly in the molecular range there is always the difficulty that the analysis method is not affecting the result, especially in vivo. It is also a challenge to study interactions between molecular inhibitors and amyloid peptides [95]. Using a series of experimental approaches, such as X-ray diffraction, nuclear magnetic resonance, scanning probe microscopy, and electron microscopy has shown progress and might also be developed further in the future.
There is a long list of biochemical and molecular biological methods to analyse protein aggregates. Depending on the question that one wants to answer different methods must be used and should be combined in an intelligent way.
Protein aggregates are a heterologous group of interacting peptide or protein subunits that can be unspecifically bound together or highly ordered. They are not fixed but can reassemble from monomers to oligomers and higher aggregates. They also interact with other components like membranes of the cell. They occur in different forms in vivo in healthy organisms and in diseases and in vitro.
For amyloids that were first found in connection with diseases like prion diseases or Alzheimer, meanwhile different function like storage and virulence were postulated. These functions of amyloid include also structure and protection, epigenetic inheritance and memory has been described for prions in mammals and yeast [96].
On the structural basis, many details have been analysed by now. Most important is the cross-β fold that occurs in different variations. This structure grows by recruitment of the corresponding amyloid protein. Knowledge of the structure facilitates understanding of function. In amyloids the repetitiveness can explain an activity that could not be executed by a monomer.
Structural aggregates like shells of viruses, microtubules, collagen, muscles and tendons are long known. For amyloids functions are meanwhile recognized and the investigation of these protein aggregates is a broad field of research now. Combining the knowledge of old methods and newly established techniques will lead to further advancement in understanding proteins.
I thank Rudi Glockshuber and Jeremias Kaegi for help with the enumeration of analytical methods and fruitful discussions and the latter additionally especially for his helpful books and proof-reading and correction of part of the manuscript.
- Fink A. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des. 1998;3:R9-23 pubmed
- Rambaran R, Serpell L. Amyloid fibrils: abnormal protein assembly. Prion. 2008;2:112-7 pubmed
- Sipe J, Cohen A. Review: history of the amyloid fibril. J Struct Biol. 2000;130:88-98 pubmed
- Wickner R, Taylor K, Edskes H, Maddelein M, Moriyama H, Roberts B. Prions in Saccharomyces and Podospora spp.: protein-based inheritance. Microbiol Mol Biol Rev. 1999;63:844-61, table of contents pubmed
- Stefani M, Dobson C. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med (Berl). 2003;81:678-99 pubmed
- Meredith S. Protein denaturation and aggregation: Cellular responses to denatured and aggregated proteins. Ann N Y Acad Sci. 2005;1066:181-221 pubmed
- Litvinov R, Gorkun O, Owen S, Shuman H, Weisel J. Polymerization of fibrin: specificity, strength, and stability of knob-hole interactions studied at the single-molecule level. Blood. 2005;106:2944-51 pubmed
- Gabizon R, McKinley M, Prusiner S. Purified prion proteins and scrapie infectivity copartition into liposomes. Proc Natl Acad Sci U S A. 1987;84:4017-21 pubmed
- Liu J, Andya J, Shire S. A critical review of analytical ultracentrifugation and field flow fractionation methods for measuring protein aggregation. AAPS J. 2006;8:E580-9 pubmed
- Schrödel A, de Marco A. Characterization of the aggregates formed during recombinant protein expression in bacteria. BMC Biochem. 2005;6:10 pubmed
- Hong P, Koza S, Bouvier E. Size-Exclusion Chromatography for the Analysis of Protein Biotherapeutics and their Aggregates. J Liq Chromatogr Relat Technol. 2012;35:2923-2950 pubmed
- Rosenberg A. Effects of protein aggregates: an immunologic perspective. AAPS J. 2006;8:E501-7 pubmed
- Carpenter J, Randolph T, Jiskoot W, Crommelin D, Middaugh C, Winter G. Potential inaccurate quantitation and sizing of protein aggregates by size exclusion chromatography: essential need to use orthogonal methods to assure the quality of therapeutic protein products. J Pharm Sci. 2010;99:2200-8 pubmed publisher
- Giddings J. Field-flow fractionation: analysis of macromolecular, colloidal, and particulate materials. Science. 1993;260:1456-65 pubmed
- Philo J. Is any measurement method optimal for all aggregate sizes and types?. AAPS J. 2006;8:E564-71 pubmed
- Fancy D, Kodadek T. Chemistry for the analysis of protein-protein interactions: rapid and efficient cross-linking triggered by long wavelength light. Proc Natl Acad Sci U S A. 1999;96:6020-4 pubmed
- Bitan G, Lomakin A, Teplow D. Amyloid beta-protein oligomerization: prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J Biol Chem. 2001;276:35176-84 pubmed
- Demeule B, Gurny R, Arvinte T. Detection and characterization of protein aggregates by fluorescence microscopy. Int J Pharm. 2007;329:37-45 pubmed
- Bertoncini C, Celej M. Small molecule fluorescent probes for the detection of amyloid self-assembly in vitro and in vivo. Curr Protein Pept Sci. 2011;12:205-20 pubmed
- Nelson R, Eisenberg D. Structural models of amyloid-like fibrils. Adv Protein Chem. 2006;73:235-82 pubmed
- Astbury W, Dickinson S, Bailey K. The X-ray interpretation of denaturation and the structure of the seed globulins. Biochem J. 1935;29:2351-2360.1 pubmed
- Sunde M, Blake C. The structure of amyloid fibrils by electron microscopy and X-ray diffraction. Adv Protein Chem. 1997;50:123-59 pubmed
- Ohnesorge F, Binnig G. True atomic resolution by atomic force microscopy through repulsive and attractive forces. Science. 1993;260:1451-6 pubmed
- Hansma H, Hoh J. Biomolecular imaging with the atomic force microscope. Annu Rev Biophys Biomol Struct. 1994;23:115-39 pubmed
- Jaroniec C, MacPhee C, Bajaj V, McMahon M, Dobson C, Griffin R. High-resolution molecular structure of a peptide in an amyloid fibril determined by magic angle spinning NMR spectroscopy. Proc Natl Acad Sci U S A. 2004;101:711-6 pubmed
- Benjwal S, Verma S, Röhm K, Gursky O. Monitoring protein aggregation during thermal unfolding in circular dichroism experiments. Protein Sci. 2006;15:635-9 pubmed
- Braun A, Alsenz J. Development and use of enzyme-linked immunosorbent assays (ELISA) for the detection of protein aggregates in interferon-alpha (IFN-alpha) formulations. Pharm Res. 1997;14:1394-400 pubmed
- Pan T, Chang B, Wong P, Li C, Li R, Kang S, et al. An aggregation-specific enzyme-linked immunosorbent assay: detection of conformational differences between recombinant PrP protein dimers and PrP(Sc) aggregates. J Virol. 2005;79:12355-64 pubmed
- Kopito R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524-30 pubmed
- Materials and Methods [ISSN : 2329-5139] is a unique online journal with regularly updated review articles on laboratory materials and methods. If you are interested in contributing a manuscript or suggesting a topic, please leave us feedback.