DNA methylation is a chemical post-replicative modification affecting CG dinucleotides, in which a methyl group (CH3) is added to these dinucleotides in a covalent manner [4]. Other forms of DNA methylation exist, for example, N6-methyladenine (6mdA) in prokaryotes [5]. DNA methylation is catalyzed by DNA methyltransferase enzymes (Dnmts) that takes place at the carbon 5 of the cytosine ring of 5’ to 3’ oriented CG dinucleotides, known as CpG sites [6] (Figure 1). Methylation in CpG dinucleotides is variable and patterns of DNA methylation can be distinguished along the genome [6-8]. Regions with a high frequency of CpG sites are known as CpG islands and are usually found in gene promoters [9]. Because the process of DNA methylation involves the enzymatic addition of methyl groups to CpG dinucleotides, it requires the availability of methyl-donor compounds. Methyl groups derive from the diet, with common sources being folic acid, betaine and vitamin B12. The availability of these compounds ultimately influences the metabolism of methionine and S-adenosyl methionine (SAM) [10], which is formed from methyl groups and is the primary methyl donor for several methyltransferases within the organism [11]. Several DNA methyltransferase enzymes (Dnmts) are currently known, each with distinct but sometimes overlapping roles. Dnmt3A and Dnmt3B are involved in the establishment of DNA methylation patterns on unmethylated DNA during early development, therefore acting as de novo methyltransferases [12]. Dnmt1, in turn, maintains these DNA methylation patterns, acting preferentially on hemi-methylated DNA [13]. The DNA methylation machinery has been identified in algae, fungi, plants, invertebrates and vertebrates [14]. Cytosine modification changes in a circadian manner in the mouse liver and lung [15]. Researchers have started to use DNA methylation profiles to classify central nervous system tumors [16, 17], to clock the ageing process [18], and, even, to reconstruct Denisovan anatomical features [19].

The main known mechanism by which DNA methylation can regulate gene expression is by preventing the binding of factors to the DNA that promotes transcriptional activity [20], thereby repressing gene expression. However, DNA methylation can also induce gene expression by preventing the binding of insulators that repress gene expression, allowing for gene expression to take place induced by the activity of nearby enhancers [21, 22].
Critical periods in which DNA methylation patterns are particularly susceptible to reprogramming exists. The main periods include the period from fecundation to blastocyst preimplantation, and also early germline differentiation [23]. However, other sensitive periods have also been described [24]. Although epigenetic programming in utero has traditionally been thought to be irreversible, a recent study has shown that epigenetic changes mediated by the maternal diet can be reversed after folic acid supplementation in the juvenile-pubertal period [25]. The mechanism by which this occurs is yet to be determined.
During the developmental initiation of the germline major epigenetic erasure occurs [23, 26], followed by reestablishment of DNA methylation patterns at the time of initiation of sex determination [27, 28]. In addition to this resetting in DNA methylation, major changes in other epigenetic mechanisms also occur during primordial germ cell development [29]. This major epigenetic reprogramming period represents a window of sensitivity to environmental factors, when new epigenomic conformations can be induced and further perpetuated across generations [30, 31].
In recent years it became clear that an interplay of DNA-methylation and DNA demethylation events is necessary for epigenetic regulation during development [32, 33].
This mechanism of adult disease etiology in which developmental epigenetic processes are considered is defined as ‘developmental origin of diseases’ [34, 35]. A variety of diseases have recently been found to have a crucial epigenetic component, including allergies [36], hepatic cancer [37], gastric cancer [38], asthma [39], colorectal cancer [40], prostate cancer [41], HIV latency [42], metabolic diseases [43, 44] and cardiovascular diseases [45]. Other studies have described the association of DNA methylation with maternal effects and related socio-biological aspects such as behavior [46], depression [47] and brain disorders [48]. Given the increasing evidence showing correlations between DNA methylation and diseases, the use of informative and reliable methods to measure DNA methylation is paramount.
Improvements in the technology to investigate epigenetic marks have been substantial in the past 30 years and have allowed a significant advance in understanding the role of epigenetics in medicine and biology in general. Current epigenetic methods can be separated into three categories: global methylation, local methylation and genome-wide methylation. Global methylation analyses were the first methods developed and they focused on determining total levels of DNA methylation in a genome, in spite of regional changes in DNA methylation. Methods to evaluate global methylation involved measurement of the incorporation of radioactive methyl groups from the S-adenosyl-L-methionine (SAM) donor into a DNA sample catalyzed by the Sss1 methylase enzyme [49, 50]. Other methods took advantage of the differential activity of some restriction enzymes depending on the methylation status of restriction sites that include CpG sites [51, 52]. More recent methods developed to measure global methylation include fluorescent quantification of DNA methyl groups using binding to anti-methyl cytosine antibodies [53, 54], high performance liquid chromatography detection of methylated cytosines [55-57] coupled with either UV (HPLC-UV) [58] or tandem mass spectrometry (LC-MS/MS) analysis [59, 60], ELISA( enzyme-linked immunosorbent assay)-based methods [61], and determination of methylation in repeat element as a representation of the methylation status of the genome, using short repeats [62] or line elements [63] in which 5mCs are detected either by a biotin-streptavidin immobilization [64], ELISA or pyrosequencing [65]. A limitation to global methylation analyses is that they can only detect major DNA methylation changes, ignoring local changes. K Douvlataniotis et al discussed drawbacks for antibody, MS, and SMRT-seq approaches in mis-identifying 6mdA (false positives) in mammals [5]. However, local changes in methylation are crucial because the majority of effects derived from exposures to compounds will occur at a local level of methylation rather than having an effect at the level of the whole genome.
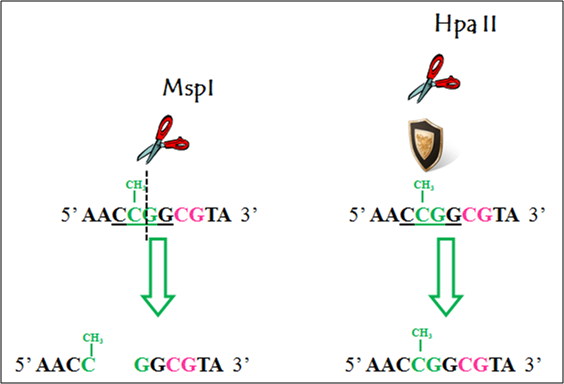
Local methylation analyses involve the determination of DNA methylation patterns of specific genes or genomic regions. Initially, both PCR amplification [66] and Southern blot detection [51] were used after methylation-sensitive restriction enzyme digestion to determine the methylation status of specific genes or regions. One common pair of restriction enzymes used for determining DNA methylation differences is HpaII and its isoschizomer MspI [51, 52, 67]. Both of these restriction enzymes cut on the CG dinucleotide of the CCGG restriction site. However, MspI is methylation-insensitive, meaning that it will cut the CG dinucleotides in spite of its methylation status. HpaII, in turn, will not cut if the CG dinucleotide is methylated (Figure 2). However, the most used method to detect local DNA methylation changes, which has the resolution at the level of CpG sites, is the Bisulfite Sequencing. The method was initially described by Frommer et al [68] and further optimized in more recent publications [69, 70]. The method involves the differential conversion of cytosine or methylated cytosine to uracil when DNA is treated with sodium bisulfite. Further PCR amplification and sequencing allow for this differential conversion to be detected. While methylated cytosines are sequenced as cytosines, unmethylated cytosines are sequenced as thymines (Figure 3). Different detection techniques can be used after bisulfite conversion, including digestion by restriction enzymes followed by PCR (Combined Bisulfite Restriction Analysis, COBRA) [71, 72], direct sequencing [73], cloning and sequencing [74], pyrosequencing [75] or mass spectrometry analysis [76]. Programs for the design of methylation-specific primers are available online, e.g., Methprimer (http://www.urogene.org/methprimer, [77] ). Analysis of the methyl-specific PCR products can be done either based on their melting profiles or the cycle threshold value (Ct value) if real-time PCR is used [78, 79]. A variation of the PCR-based detection method, COLD-PCR, uses low denaturation temperature to allow the preferential amplification of the unmethylated DNA fragments [80]. Direct sequencing can measure several DNA molecules at the same time. Nevertheless, problems in the sequencing signals arise due to the major overrepresentation of thymine signals versus cytosine signals after the bisulfite conversion. Moreover, sequencing software is not devoted to quantifying sequencing peaks, rather, they present normalized signals that do not represent quantification of two overlapping signals in a particular CpG peak. Because of this, direct sequencing measurements of DNA methylation can be distorted. Corrections to these problems have involved either the use of algorithms or complementary experimental procedures. An algorithm has been described that compensates the overrepresentation of the thymine over the cytosine signal by normalizing the signals from the other nucleotides with the thymine signal [81]. An algorithm has been described that compensates the overrepresentation of the thymine over the cytosine signal by normalizing the signals from the other nucleotides with the thymine signal [81], and most recently, a complete bisulfite sequencing analysis workflow became available [82]. Also, bisulfite conversion followed by cloning corrects these issues, by producing clean and defined electropherograms that result from the sequencing of clones representing the methylation status of only one initial molecule [70]. Nevertheless, the number of molecules analyzed will depend on the number of clones and the error associated can be drastic if only a few clones are measured. Digital bisulfite sequencing allows for the simultaneous interrogation of DNA methylation states of an increased number of single DNA molecules, by substituting the process of cloning by dilutions of DNA that also produce amplification of single molecules [83]. Although pyrosequencing [75] produces quantifiable signals per CpG being measured, it has the disadvantage of allowing for only short regions to be evaluated per amplicon, in average 150 bp in size, which limits the span of CpGs to be analyzed. Bisufite treatment followed by mass spectrometry detection [76] allows for amplicons of increased size (up to approximately 500 bp) to be interrogated.
A new, highly accurate method to detect DNA methylation through bisulfite sequencing (HAM-TBS) has also been developed for the analysis of FKBP5 gene, one of the regulatory genes involved in the stress pathway [84]. Another study has presented a method, which mingled hairpin bisulfite and oxidative bisulfite sequencing to detect the main DNA methylation variants in mouse embryonic stem cells and to predict the activity of oxidation Tet enzymes [85]. In addition, a new technique utilizing an HPLC anion-exchange column with hydrophobic and electrostatic characteristics has reported, in which the genomic DNA undergoes bisulfite treatment, followed by HPLC separation and quantification of DNA methylation [86].
Various bisulfite treatment kits, through digital PCR, qPCR, QuARTS [87] or its derivatives like LQAS [88], or electrophoresis, for the detection of DNA methylation are currently available. Kint et al compared the effectiveness of several commercial bisulfite-based kits and suggested that digital PCR was a more reliable approach to evaluate the methylation [89]. Bisulfite treatment-based sequencing on single cells (single-cell DNA methylome profiling) is also optimized (snmC-seq, snmC-seq2) [90].

An electrophoresis-based method for the separation of methylated DNA is also available [91]. The method requires the use of denaturing-gradient polyacrylamide gels and it is time-consuming and labor-intensive. Also, a recent study has used dot blot assay, where methylation of genomic DNA was analyzed by placing the genomic DNA on an N+ (positively charged nylon) membrane, and DNA methylation was detected through a commercial antibody to 5-methylcytosine (5-mC) [92].
A rapid colorimetric assay using the methyl-binding domain (MBD) proteins for the evaluation of total genomic and gene-specific methylation from low amounts of DNA has been recently developed [1]. With this approach, genomic DNA is digested with restriction enzymes, biotinylated via a fill-in reaction with Klenow polymerase and biotin-dNTPs, and selected with MBD magnetic beads. Enriched, biotinylated methyl-DNA is then evaluated by a streptavidin-HRP (horseradish peroxidase) mediated reaction (Figure 4). To detect gene-specific methylation, gene amplification is performed with biotin-dNTP and the generated biotin-DNA polymers are selected and evaluated with SA magnetic beads or SA-HRP. The assay is also adapted for electrochemical detection of the methylated DNA.
In addition, DNA-linked silver nanoclusters have also been used for the analysis of DNA methylation [93]. This technique detects both DNA methylation and activity of DNA methyltransferase and evaluates the changes in the fluorescence intensity induced by methylated cytosine residues.

The first methods described to interrogate gene- or region-specific DNA methylation changes on a genome-wide scale were also based on the differential sensitivity to restriction enzymes to cut methylated or unmethylated restriction sites. The AIMS (Amplification of Inter-Methylated Sites) method uses a combination of the methylation-sensitive SmaI and its isoschizomer methylation-insensitive PspAI enzyme to construct fragments to which adaptors are ligated to perform a further whole-genome PCR reaction [94]. Differential amplification of fragments then indicates changes in DNA methylation. A similar approach is taken by the HELP (HpaII tiny fragments Enrichment by Ligation-mediated PCR) assay, which uses the methylation-sensitive HpaII restriction enzyme and its isoschizomer methylation insensitive MspI enzyme [67].
One of the most powerful approaches described to interrogate for regional DNA methylation changes at a genome-wide scale is based on the chromatin immune-precipitation of methylated fragments (MeDIP) [95]. With this method methylated DNA is enriched from an initial DNA sample through immuno-precipitation with an anti-methylcytosine antibody (MeDIP). DNA samples enriched for methylated DNA are then hybridized to microarray chips (MeDIP-Chip) for identification of the locations with DNA methylation [95]. This tool has been used to map the methylome in Arabidopsis thaliana [96], monoamine oxidase A and B regions [97], human breast cancer metastasis [98] and the human major histocompatibility complex [99]. MeDIP-Chip has the obvious advantage of being able to scan for methylation changes in the whole genome. However, false positives can arise, making further confirmation of the sites with the local methylation tools previously described needed after the genome-wide detection. Another array-based method to interrogate genome-wide changes in DNA methylation takes advantage of the restriction activity by the methylation-dependent enzyme McrBC in order to deplete the methylated DNA fraction and compare with an intact fraction, when hybridizing to chip arrays [100]. More recently, the use of the Infinium HumanMethylation27 Bead-Chip has specifically focused on regions of interest for DNA methylation change that could correlate with diseases in humans [101, 102]. This method is an adaptation of the Golden Gate SNP genotyping assay that uses bisulfite converted DNA. Therefore, SNPs of interest are C/T conversions that represent CpG methylation changes.

A major extension of Infinium HumanMethylation27 Bead Array, Infinium HumanMethylation450 (HM450), became the most used method for DNA methylation profiling in recent years [103, 104]. The array can determine the methylation status of ~485,000 CpGs in coding and non-coding DNA regions, including miRNA promoters, and 5’ and 3’ UTRs in human DNA. The Illumina Infinium Methylation EPIC kit can obtain the DNA methylation status at >850,000 CpG sites and is very popular [105]. Lambo S et al clustered both FFPE and fresh-frozen ETMR samples based on HM450 DNA methylation profiling [103]. Coorens THH et al obtained the genome-wide methylation profiles of normal kidneys and kidney cancer tissues with Illumina Infinium MethylationEPIC BeadChip microarray kit [106]. Venkataramani V et al characterized glioblastoma stem cell lines with the Illumina Infinium MethylationEPIC kit [16]. Illumina TruSeq Methyl Capture EPIC Library Prep Kit can interrogate over 3.3 million CpG sites [59].
MeDIP has also been coupled with sequencing in order to use a reduced representation of the genome, the methylated genome, as input for sequencing (MeDIP-seq) [107]. Another method has been described to capture the methylated fraction of DNA by methyl-binding proteins (MeCP2) followed by sequencing, named the MethylCap-seq technique [108]. Also, the treatment of DNA with MspI has been used to reduce the genome to about 1% of its total, producing a fraction of interest that contains the majority of CpG sites [109, 110]. This DNA fraction is then converted with bisulfite treatment and sequenced. This method is called Reduced Representation Bisufite Sequencing (RRBS) [110]. For example, DiTroia SP et al identified 460 differentially methylated regions between vitamin-C-depleted and control mouse E13.5 germ cells with RRBS [111]. A comparison between the MeDIP-seq, MethylCap-seq and RRBS methods concludes that all produce accurate DNA methylation data, although MeDIP-seq and MethylCap-seq provide broader coverage of the genome whereas RRBS has limited coverage on CpG poor regions [112]. Also, MethylCap-seq is able to identify more differentially methylated regions (DMR) than RRBS, which in turn identifies more DMR than MeDIP-seq [112].
Nevertheless, notable variability of genomic data obtained from DIP-seq research has been observed. A recent study by Lentini et al has found that all antibodies, which are usually applied for DIP-seq, bind nonspecific STR sequences [113]. The authors have demonstrated that up to 99% observed DNA methylation in publications might be caused by off-target binding. Therefore, DIP-seq studies must use an IgG control [113]. Similar non-specific binding problems are also identified for anti-6mdA antibodies [5].
A more recent comparison between MethylCap-seq and Illumina's Infinium HumanMethylation450 BeadChips (HM450) [114] indicates that the two methods are complementary, with HM 450 being more sensitive, while MethylCap-seq having a much larger genome-wide coverage.
A more recent and very promising tool that is able to overcome limitations related to accurately quantify DNA methylation at the CpG resolution on a genome-wide level is deep sequencing, which has been used to map the methylome in Arabidopsis [115] and humans [116]. This method involves bisulfite conversion of unmethylated cytosines followed by adapter ligation and whole genome sequencing. However, the high cost of the technique is still a limiting factor to select it over techniques that incorporate sequencing after reducing the genome representation to its methylated fraction. A more recent method named single-molecule, real-time sequencing (SMRT) involves incorporation of fluorescently labeled nucleotides into complementary nucleic acid strands [117]. The kinetics of this incorporation allows for the determination of several epigenetic marks, including DNA methylation and hydroxymethylation, without the need for bisulfite conversion. A summary of the pros and cons of each method described here for the analysis of DNA methylation is shown in Table 1.

During the last few years, the study of DNA demethylation became of great interest in the field of epigenetics. In mammals, DNA demethylation is achieved via TET-mediated oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxycytosine (5caC). Two main methods for their detection have been developed. One method is bisulfite-treatment independent, while the second one uses the bisulfite treatment technique combined with methyltransferase treatment of cytidines.
The bisulfite-free method detects whole-genome 5fC. The method involves chemical labeling of the 5-formylcytosine, conjugation to biotin for pull-down enrichment, removal of 5fC-free strands, and ligation to adaptors prior to PCR amplification. The obtained DNA is subjected to next-generation sequencing, and C-to-T transitions are identified to define 5fC sites in the whole genome [118].
The second method, called methylation-assisted bisulfite sequencing (MAB-seq) was developed for the detection of 5fCs and 5caCs at single nucleotide resolution. In this method, DNA is treated with the S-adenosyl-methionine-dependent CpG methyltransferase M.SssI prior to bisulfite treatment, which leads to the selective conversion of unmodified cytosines to 5-methylcytosine, and the detection of 5fC and 5caC residues (Figure 5) [2]. An improved version of the protocol called reduced-representation MAB-seq (RRMAB-seq) [119] provides increased coverage on CpG-rich regions, and reduces the execution costs of the technique.
Multiple commercial kits can be integrated to simplify the DNA methylation quantitation. For example, Gkountela S et al performed proteinase K digestion and bisulfite treatment of individual CTC cells or clusters with Zymo EZ DNA Methylation Direct Kit (Cat# D5020), generated a library with Illumina TruSeq DNA methylation kit (Cat# EGMK91396), amplified the library with Illumina Failsafe Enzyme (Cat# FSE51100), introduced indexes with Illumina Index Primers’ Kit (Cat# EGIDX81312), purified the library with Agencourt AMPure XP beads, and estimated the library concentration with Invitrogen Qubit DS DNA HS Assay Kit (Cat#Q32854) [120]. Zymo EZ DNA Methylation Direct Kit is a popular choice for bisulfite amplicon sequencing [59].

AbaSI-coupled sequencing [121] uses the restriction endonuclease AbaSI that selectively binds 5gmCs, but not 5mCs or Cs and generates double-stranded DNA breaks at the 3’ end of binding sites. AbaSI preferred sites are shown in Figure (Figure 6A). In this method, genomic 5hmCs are converted to 5gmCs by T4 β-glucosyltransferase, digested with AbaSI and further ligated to biotinylated double-stranded adaptors containing a random 3’-overhang. DNA is captured on beads and ligated to a dT-based adaptor that facilitates dA-tailing. Fragments are then amplified by PCR, prior to sequencing (Figure 6B).
By adapting the Aba-seq technique for 5hmC sequencing, Mooijman et al [122] developed a method for genome-wide detection of 5hmCs in single cells sorted on 384-well plates. In this version of the technique, the researchers replaced the biotinylated adaptor with one that contains a cell-specific barcode, the Illumina 5’-adaptor and a T7 promoter. The glycosylated DNA was transcribed in vitro for sequence amplification purposes. The obtained RNA was fragmented and included in RNA sequencing libraries (Figure 7).
| Category | Method description | Pros | Cons | References |
|---|---|---|---|---|
| Global methylation analysis | Radioactive incorporation of methyl groups | Inexpensive; Complete coverage of CpG sites | Not suitable to measure local methylation changes; Radioactively based | [49, 50] |
| Combination of restriction enzymes with differential sensitivity to DNA methylation | Inexpensive; Non-radioactive; Requires basic instrumentation | Not suitable to measure local methylation changes; Representative but not complete coverage of CpG sites | [51, 52] | |
| Fluorescent quantification of DNA methyl groups captured by anti-methylcytosine antibody | Non-radioactive; Measures all CpG sites | Not suitable to measure local methylation changes; Costs associated with specific instrumentation and antibody | [54, 123] | |
| HPLC detection of methyl-cytosines | Non-radioactive; Measures all CpG sites | Not suitable to measure local methylation changes; Costs associated with specific instrumentation; Advanced knowledge of instrumentation | [55-57] | |
| Determination of DNA methylation in repeat elements | Non-radioactive; Requires only basic instrumentation | Not suitable to measure local methylation changes; Representative but not complete coverage of CpG sites | [62, 63] | |
| LC-MS/MS | Non-radioactive; requires small quantities of DNA | Requires advanced training and special instrumentation | [60]||
| ELISA | Non-radioactive; Requires only basic instrumentation | Costs associated with specific antibody | [61] | |
| Biotin-SA-HRP-mediated colorimetric assay | Non-radioactive; rapid; requires low amounts of DNA | Requires biotin-dNTPs and the use of magnetic beads | [1] | |
| Local methylation analysis | PCR amplification of DNA material digested with restriction enzymes differentially sensitive to CpG DNA methylation | Non-radioactive; Requires only basic instrumentation | Coverage limited to CpG sites target of restriction; | [66] |
| Southern blot detection of genomic regions from DNA material digested with restriction enzymes differentially sensitive to CpG DNA methylation | Requires only basic instrumentation | Radioactive blotting; Coverage limited to CpG sites target of restriction. | [51] | |
| Bisulfite conversion and direct sequencing | Requires only basic instrumentation; Capable of detecting DNA methylation at CpG resolution independent of restriction site limitations | Primer design is sometimes difficult due to long stretches of thymines after bisulfite conversion. Electropherogram sequencing signals are difficult to quantify due to the overrepresentation of thymines over cytosines. | [68, 73] | |
| Bisulfite conversion and PCR amplification of DNA material digested with restriction enzymes differentially sensitive to CpG to TpG conversion (COBRA) | Very simple method that requires only basic instrumentation. | Primer design is sometimes difficult due to long stretches of thymines after bisulfite conversion; Coverage limited to CpG sites target of restriction. | [71, 72] | |
| Bisulfite conversion, cloning and sequencing | Requires only basic instrumentation; Capable of detecting DNA methylation at CpG resolution independent of restriction site limitations; Clean sequencing electropherograms are generated. | Primer design is sometimes difficult due to long stretches of thymines after bisulfite conversion. High numbers of clones per sample are required to produce comparisons with enough statistical power. | [74, 124] | |
| Bisulfite conversion, dilution and PCR (Digital bisulfite sequencing) | Requires only basic instrumentation; Capable of detecting DNA methylation at CpG resolution independent of restriction site limitations; Clean electropherograms. | Primer design is sometimes difficult due to long stretches of thymines after bisulfite conversion | [83] | |
| Bisulfite conversion and pyrosequencing | Requires only basic instrumentation; Capable of detecting DNA methylation at CpG resolution independent of restriction site limitations; Very quantifiable. | Limited possibilities of amplicon size; Primer design is sometimes difficult due to long stretches of thymines after bisulfite conversion | [75] | |
| Bisulfite conversion and mass spectrometry detection | Capable of detecting DNA methylation at CpG resolution independent of restriction site limitations; Allows for analysis of larger amplicons than pyrosequencing | Primer design is sometimes difficult due to long stretches of thymines after bisulfite conversion; Costs associated with specific instrumentation; Advanced knowledge of instrumentation | [76] | |
| COLD-PCR | Non-radioactive; Requires only basic instrumentation | Coverage limited to CpG sites target of restriction | [80] | |
| Electrophoresis-based DNA separation | Non-radioactive; Requires only basic instrumentation | Uses denaturing-gradient polyacrylamide gels; it is time-consuming and labor-intensive | [91] | |
| Biotin-SA-HRP-mediated colorimetric assay | Non-radioactive; rapid; requires low amounts of DNA | Requires biotin-dNTPs and the use of magnetic beads | [1] | |
| Genome-wide methylation analysis | Amplification of inter methylated sites (AIMS) obtained with a combination of methyl-sensitive and methyl-insensitive enzymes | Requires only basic instrumentation; Capable of detecting regional methylation changes at a genome-wide scale. | Not a method for epigenome mapping, but rather to detect epigenetic differences; Coverage limited to CpG sites target of restriction. | [94] |
| HpaII tiny fragments enrichment by ligation mediated PCR (HELP). Enrichment is obtained with a combination of methyl-sensitive and methyl-insensitive enzymes | Requires only basic instrumentation; Capable of detecting regional methylation changes at a genome-wide scale. | Not a method for epigenome mapping, but rather to detect epigenetic differences; Coverage limited to CpG sites target of restriction. | [67] | |
| Methylated DNA immunoprecipitation and microarray chip hybridization (MeDIP-Chip) | Capable of detecting regional methylation changes at a genome-wide scale; Coverage is independent of restriction site limitations. | Costs associated with a specific antibody and with array hybridization related instrumentation; Amount of data generated requires bioinformatic knowledge; Detect regional changes in DNA methylation but without CpG resolution | [95] | |
| Depletion of the methylated fraction of DNA with the McrBC enzyme, and hybridization to chip arrays. | Capable of detecting regional methylation changes at a genome-wide scale; Coverage is independent of restriction site limitations, since MrcBC acts on all methylated cytosines. | Costs associated with array hybridization related instrumentation; Amount of data generated requires bioinformatic knowledge. | [100] | |
| Infinium HumanMethylation27 Bead-Chip assays. SNP array with bisulfite converted DNA that detects C/T SNPs (CpG methylation sites) of interest. | Capable of detecting regional methylation changes at a genome-wide scale; Coverage is independent of restriction site limitations; Efficient method to interrogate CpG sites in regions of biological relevance in humans | Costs associated with array hybridization related instrumentation; Limited to human genome; Coverage limited to a specific number of CpG sites; Amount of data generated requires bioinformatic knowledge. | [101, 102] | |
| Methylated DNA immunoprecipitation followed by sequencing (MeDIP-Seq) | Capable of detecting regional methylation changes at a genome-wide scale with CpG resolution; Coverage is independent of restriction site limitations; Costs of sequencing are not high due to the reduced representation of the genome. Good coverage of the methylome | Costs associated with a specific antibody and with sequencing; Amount of data generated requires bioinformatic knowledge. Less powerful than equivalent methods to identify differentially methylated regions. | [107] | |
| Capture of methylated DNA using the methyl DNA binding domain MeCP2 followed by sequencing (MethylCap-seq) | Capable of detecting regional methylation changes at a genome-wide scale with CpG resolution; Coverage is independent of restriction site limitations; Costs of sequencing are lower due to the analysis of a reduced representation of the genome; Good coverage of the methylome. | Costs associated with a specific antibody and with sequencing; Amount of data generated requires bioinformatic knowledge. | [108] | |
| Reduced representation bisulfite sequencing (RRBS) | Capable of detecting regional methylation changes at a genome-wide scale with CpG resolution; Coverage is independent of restriction site limitations; Costs of sequencing are lower due to the analysis of a reduced representation of the genome. | Costs associated with sequencing; Amount of data generated requires bioinformatic knowledge. Limited coverage of the methylome in CpG poor regions. | [109, 110] | |
| Deep sequencing of bisulfite treated DNA | Capable of detecting regional methylation changes at a genome-wide scale and CpG resolution; Coverage is independent of restriction site limitations; Directly quantifiable detection of DNA methylation on CpG sites. | High costs associated with sequencing; Amount of data generated requires bioinformatic knowledge. | [125] | |
| Single molecule, real-time sequencing (SMART) | Capable of detecting regional methylation changes at a genome-wide scale and CpG resolution; Coverage is independent of restriction site limitations; Directly quantifiable detection of DNA methylation on CpG sites; No need of bisulfite conversion. | High costs associated with sequencing; Amount of data generated requires bioinformatic knowledge. | [117] | |
| Infinium HumanMethylation45 and the VeraCode Methylation Array from Illumina | Extension of Infinium HumanMethylation27 Bead Array; increased number of CpG sites covered. | Costs associated with array hybridization related instrumentation; Limited to human genome; A large amount of data generated requires bioinformatic analysis. | [114, 126] | |
| DNA methylation dynamics | Bisulfite-free detection of 5fC | Non-radioactive; PCR-based amplification and NGS. | Chemical-labeling reaction the efficiency problems; requires advanced skills. | [118] |
| Methylation-assisted bisulfite sequencing (MAB-seq) and reduced-representation MAB-seq (RRMAB-seq) | Detection of 5fCs and 5caCs at single nucleotide resolution. | Depends on the efficiency of DNA methylation reaction; high execution costs. | [2, 119] | |
| 5hmC-Seal / Aba-seq and variants | Genome-wide detection of 5hmCs at single nucleotide resolution. Adapted to detection of 5hmCs in single cells. | Depends on the efficiency of 5hmCs glycosylation. | [59, 121, 122] |
Reliable and quantifiable analysis of DNA methylation patterns is becoming paramount due to the recent advances in epigenetic research and the association of altered methylation patterns with several non-infectious diseases of common occurrence. The technology to determine DNA methylation changes has seen steady progress in the past 30 years. Initial methods included global measures of DNA methylation that overlooked important local changes in DNA methylation. Further developments, however, led to reliable and quantifiable analysis of DNA methylation in specific genes or genomic regions. Current methods have expanded this capability to reliably interrogate DNA methylation changes with the region or CpG specific precision at a genome-wide scale. Due to the recent boom in the development of sequencing technologies and the consequent reduction in cost to perform sequencing assays, DNA methylation analyses based on sequencing technology are becoming accessible to an increasing number of laboratories around the world. In the near future, it is expected that the sequencing costs will be further reduced, allowing for quantifiable interrogation of DNA methylation at all CpG sites in the genome. However, choosing the right method for a specific project might still be challenging. A recently published comparative review of DNA methylation analysis techniques [3] includes an algorithm for choosing appropriate experimental methods (Figure 8).

Additional challenges are created by the massive amount of data generated. Bioinformatic analyses are becoming indispensable for epigenetic studies, especially when the focus is on a genome-wide scale. The main focus of epigenetic studies is currently the molecular mechanisms of diseases, and developmental and reproductive biology. Reduction in cost for DNA methylation assays will allow for consolidation of DNA methylation analysis in these fields, as well as for the expansion of DNA methylation analyses to other fields of biology and to organism models not currently analyzed. Also, reduced assay costs will allow for increased numbers of individuals to be used in DNA methylation research, which will produce better data and epigenetic comparisons.
- Laird P, Jaenisch R. The role of DNA methylation in cancer genetic and epigenetics. Annu Rev Genet. 1996;30:441-64 pubmed
- Singal R, Ginder G. DNA methylation. Blood. 1999;93:4059-70 pubmed
- Bestor T. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395-402 pubmed
- Jones P, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068-70 pubmed
- Gardiner Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261-82 pubmed
- Van den Veyver I. Genetic effects of methylation diets. Annu Rev Nutr. 2002;22:255-82 pubmed
- Yokochi T, Robertson K. Preferential methylation of unmethylated DNA by Mammalian de novo DNA methyltransferase Dnmt3a. J Biol Chem. 2002;277:11735-45 pubmed
- Yoder J, Soman N, Verdine G, Bestor T. DNA (cytosine-5)-methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. J Mol Biol. 1997;270:385-95 pubmed
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089-93 pubmed
- Edwards T, Myers J. Environmental exposures and gene regulation in disease etiology. Environ Health Perspect. 2007;115:1264-70 pubmed
- Lees Murdock D, Walsh C. DNA methylation reprogramming in the germ line. Epigenetics. 2008;3:5-13 pubmed
- Allegrucci C, Thurston A, Lucas E, Young L. Epigenetics and the germline. Reproduction. 2005;129:137-49 pubmed
- Durcova Hills G, Hajkova P, Sullivan S, Barton S, Surani M, McLaren A. Influence of sex chromosome constitution on the genomic imprinting of germ cells. Proc Natl Acad Sci U S A. 2006;103:11184-8 pubmed
- Godfrey K, Lillycrop K, Burdge G, Gluckman P, Hanson M. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr Res. 2007;61:5R-10R pubmed
- Pogribny I, Ross S, Wise C, Pogribna M, Jones E, Tryndyak V, et al. Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutat Res. 2006;593:80-7 pubmed
- Nan H, Song Y, Yun H, Park J, Kim H. Effects of dietary intake and genetic factors on hypermethylation of the hMLH1 gene promoter in gastric cancer. World J Gastroenterol. 2005;11:3834-41 pubmed
- Lillycrop K, Slater Jefferies J, Hanson M, Godfrey K, Jackson A, Burdge G. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation. Br J Nutr. 2007;97:1064-73 pubmed
- Champagne F, Weaver I, Diorio J, Dymov S, Szyf M, Meaney M. Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology. 2006;147:2909-15 pubmed
- Oberlander T, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin A. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97-106 pubmed
- Balaghi M, Wagner C. DNA methylation in folate deficiency: use of CpG methylase. Biochem Biophys Res Commun. 1993;193:1184-90 pubmed
- Soares J, Pinto A, Cunha C, Andre S, Barao I, Sousa J, et al. Global DNA hypomethylation in breast carcinoma: correlation with prognostic factors and tumor progression. Cancer. 1999;85:112-8 pubmed
- Goelz S, Vogelstein B, Hamilton S, Feinberg A. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science. 1985;228:187-90 pubmed
- Ben Hattar J, Jiricny J. Effect of cytosine methylation on the cleavage of oligonucleotide duplexes with restriction endonucleases HpaII and MspI. Nucleic Acids Res. 1988;16:4160 pubmed
- Friso S, CHOI S, Dolnikowski G, Selhub J. A method to assess genomic DNA methylation using high-performance liquid chromatography/electrospray ionization mass spectrometry. Anal Chem. 2002;74:4526-31 pubmed
- Ramsahoye B. Measurement of genome wide DNA methylation by reversed-phase high-performance liquid chromatography. Methods. 2002;27:156-61 pubmed
- Kuo K, McCune R, Gehrke C, Midgett R, Ehrlich M. Quantitative reversed-phase high performance liquid chromatographic determination of major and modified deoxyribonucleosides in DNA. Nucleic Acids Res. 1980;8:4763-76 pubmed
- Song L, James S, Kazim L, Karpf A. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal Chem. 2005;77:504-10 pubmed
- Kim H, Park J, Jeong K, Lee S. Determining the global DNA methylation status of rat according to the identifier repetitive elements. Electrophoresis. 2007;28:3854-61 pubmed
- Yang A, Estecio M, Doshi K, Kondo Y, Tajara E, Issa J. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:e38 pubmed
- Singer Sam J, LeBon J, Tanguay R, Riggs A. A quantitative HpaII-PCR assay to measure methylation of DNA from a small number of cells. Nucleic Acids Res. 1990;18:687 pubmed
- Khulan B, Thompson R, Ye K, Fazzari M, Suzuki M, Stasiek E, et al. Comparative isoschizomer profiling of cytosine methylation: the HELP assay. Genome Res. 2006;16:1046-55 pubmed
- Frommer M, McDonald L, Millar D, Collis C, Watt F, Grigg G, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89:1827-31 pubmed
- Clark S, Harrison J, Paul C, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990-7 pubmed
- Clark S, Statham A, Stirzaker C, Molloy P, Frommer M. DNA methylation: bisulphite modification and analysis. Nat Protoc. 2006;1:2353-64 pubmed
- Eads C, Laird P. Combined bisulfite restriction analysis (COBRA). Methods Mol Biol. 2002;200:71-85 pubmed
- Xiong Z, Laird P. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532-4 pubmed
- Paul C, Clark S. Cytosine methylation: quantitation by automated genomic sequencing and GENESCAN analysis. Biotechniques. 1996;21:126-33 pubmed
- Warnecke P, Stirzaker C, Song J, Grunau C, Melki J, Clark S. Identification and resolution of artifacts in bisulfite sequencing. Methods. 2002;27:101-7 pubmed
- Tost J, Gut I. Analysis of gene-specific DNA methylation patterns by pyrosequencing technology. Methods Mol Biol. 2007;373:89-102 pubmed
- Ehrich M, Nelson M, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A. 2005;102:15785-90 pubmed
- Li L, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427-31 pubmed
- Frigola J, Ribas M, Risques R, Peinado M. Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS). Nucleic Acids Res. 2002;30:e28 pubmed
- Weber M, Davies J, Wittig D, Oakeley E, Haase M, Lam W, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853-62 pubmed
- Ordway J, Bedell J, Citek R, Nunberg A, Garrido A, Kendall R, et al. Comprehensive DNA methylation profiling in a human cancer genome identifies novel epigenetic targets. Carcinogenesis. 2006;27:2409-23 pubmed
- Habib M, Fares F, Bourgeois C, Bella C, Bernardino J, Hernandez Blazquez F, et al. DNA global hypomethylation in EBV-transformed interphase nuclei. Exp Cell Res. 1999;249:46-53 pubmed
- Materials and Methods [ISSN : 2329-5139] is a unique online journal with regularly updated review articles on laboratory materials and methods. If you are interested in contributing a manuscript or suggesting a topic, please leave us feedback.
- method
- Antibody Quality
- CRISPR and Genomic Engineering
- Chromosome Conformation Capture
- Cloning and Expression Vectors, and cDNA and microRNA Clones Companies
- DNA Extraction and Purification
- Histone Modification
- Immunological Analysis of Chromatin and Epigenetic Modifications
- Monoclonal Antibodies: Expression and Purification in a Basic Research Laboratory
- Monoclonal Antibodies - Quality Control and Quantification through Mass Spectrometry
- Phage-Display Technology for the Production of Recombinant Monoclonal Antibodies
- Protein Modification
- RNA Extraction
- RNA Modifications
- RNA-seq
- Single Cell Technologies
- Somatic Mutations
- siRNAs and shRNAs: Tools for Protein Knockdown by Gene Silencing