An updated review of PCR methods, including conventional, real-time, microfluidic, and drop digital PCR.
The Polymerase chain reaction (PCR) is a ubiquitous technique utilized extensively for diagnostic purposes and molecular biology research. PCR is the in vitro amplification of specific nucleic acid (NA) sequences by a DNA Polymerase enzyme. The PCR technique was transformed by Kary Mullis in 1983 (who died in August 2019 at the age of 74), when he expanded the use of a heat-stable polymerase with temperature cycling [2-4]. The universal utility of PCR is that it amplifies small quantities of target nucleic acid sequences, yielding an amount of product that is detectable by downstream methods, such as visualization of NA on an agarose gel. This is due to the exponential amplification of the sequence and the resulting millions of copies of the original template.
PCR reactions amplify target nucleic acid sequences via the use of a DNA Polymerase, primers, and nucleotides. The template for a PCR reaction may be any nucleic acid sequence of interest, and the NA source may be DNA, RNA, or cDNA. Primers are short sequences of nucleotides synthesized in vitro. They are designed to anneal to opposite strands of a specific NA template target and usually are between 15-40 bases long. Primers ideally lack secondary structure and are not complementary to each other, to prevent primer-dimer formation. A variety of DNA Polymerases have been utilized for PCR, but the thermostable Taq DNA Polymerase is probably the most widely used. This enzyme adds the deoxyribonucleoside triphosphates (dNTPs or nucleotides) onto the ends of the primers to extend the NA based on the template NA sequence.
The PCR reaction mixture is temperature cycled, typically 20-40 times. Denaturation of the NA template sequence is achieved at 95ºC. The temperature is cooled to 37-60ºC to anneal primers to the target sequence. Extension of the primers with nucleotides by the DNA Polymerase is achieved at temperatures ranging from 60-72ºC. Conventional cycling conditions are 95ºC for 5 minutes initially to denature all template NA, followed by 2-40 repeats of 95ºC for 30 sec, 60ºC for 30s and 72ºC for 1 minute. The time spent at each temperature can be optimized for specific assays. For instance, the amplification of very short target sequences requires much shorter incubations at each temperature than that of very large target sequences. Each round of temperature cycling results in two times more target sequence than the prior round. This leads to the exponential amplification of the original template, often resulting in millions or billions of copies of the original NA target.
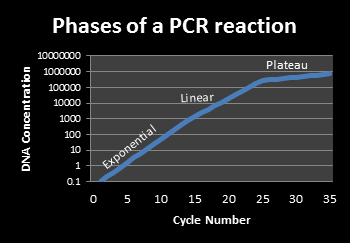
The basic PCR reaction occurs in three phases. The exponential phase is the period in which exact doubling of a nucleic acid product occurs every cycle. Real-time PCR detection is carried out during this exponential phase. The linear phase occurs as the reaction is slowing due to the consumption of the reagents and the degradation of the products. The final stage is the plateau phase, which occurs when the reaction has stopped, and no additional amplicon is being generated. This is the point at which the PCR reaction product is analyzed via gel electrophoresis for conventional PCR reactions.
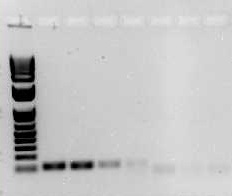
Downstream detection of the PCR product is done in many ways. A common method of visualization is via agarose gel electrophoresis. This involves separating NA fragments via electrophoresis and staining the NA with an intercalating dye such as ethidium bromide or SybrSafe and subsequent detection using a UV light source and imaging system (Figure 2).
Real-time PCR uses specialized thermocyclers that detect the fluorescent signal in each well. This signal is indicative of the amount of double-stranded NA within the reaction tube or well. This signal, in relative fluorescent units, is plotted by the thermocycler software versus cycle number (Figure 3).

Multitudes of research, clinical and forensic applications of PCR exist. In molecular biology research, PCR is often used for genetic engineering, DNA sequencing, and gene expression analyses. In clinical laboratories, PCR is crucial in the detection of infectious disease. Forensic applications of PCR include paternity testing and DNA fingerprinting. These tests benefit from the exquisite sensitivity of PCR because target sequences can be detected from a single human hair. Table 1 lists the major applications and references to manuscripts describing them.
| applications | references |
|---|---|
| detection of foodborne pathogens | [5, 6] |
| diagnosis of infectious disease | [7] |
| genotyping | [8] |
| human genetic mutation detection | [9, 10] |
| expression profiling of microRNA | [11, 12] |
| gene expression analysis | [13, 14] |
| DNA sequencing | [15, 16] |
| forensics - genetic fingerprinting | [17-19] |
| forensics - parental testing | [20] |
| genetic engineering | [21, 22] |
A wide variety of PCR methods exist, and each has advantages and limitations. Standard or conventional PCR is the most basic type of PCR reaction. It gives qualitative results and requires a post-PCR step for detection or visualization of the DNA. A major advantage to conventional PCR is the ready access to conventional thermocyclers that almost all research facilities have, and the fairly low cost. Often, conventional PCR reactions are loaded onto an agarose gel and are resolved by size via electrophoresis. The DNA is visualized by using an intercalating dye such as ethidium bromide or Sybr Safe, and a UV light source. The specificity of the PCR reaction is confirmed by size as compared to a DNA ladder, which is a mixture of known sized fragments of DNA. The bands may be isolated from the gel, and the DNA can be purified and sequenced, which is a more dependable means of specificity determination than sizing via comparison to a DNA ladder. Figure 2 gives an example of an agarose gel with a DNA ladder on the left and several PCR reactions to its right. Limitations of this type of end-point PCR include low sensitivity and non-quantitative results.
| experimental needs | PCR method | advantages | limitations |
|---|---|---|---|
| determine if a target NA sequence is present or absent in a few samples | standard or conventional | easy access to equipment minimal cost | qualitative results only post reaction handling |
| determine quantities of target NA in many samples | real-time | can generate quantitative results can be sequence specific | more expensive than conventional PCR speed is mid-level |
| determine quantities of many target NA for a few samples | PCR arrays | up to 88 genes can be measured per sample at a time | one array is needed per sample costly for many samples |
| detect target NA in the field | microfluidic chip | fast results small size portable | specialized equipment costly |
| detect very low abundance NA targets or need extremely accurate quantitation | digital and drop digital | very precise absolute quantitation | specialized equipment costly |
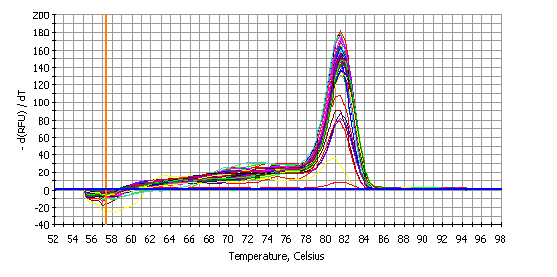
Real-time PCR, also called qPCR (quantitative PCR), is a more recent but already extremely common method of PCR that offers several advantages over conventional PCR. First, the PCR product can be detected in real time, so the need for an agarose gel to visualize the DNA post-PCR is unnecessary. Further, real-time PCR can be both quantitative and specific. Starting quantities of a sequence of interest can be determined by comparison of samples to a standard curve of known quantities of DNA. The increased specificity is achieved through the use of specific NA probes and/or a melt curve analysis that follows the PCR reaction. See Figure 4 for an example of a melt curve of a single PCR product. The presence of a single peak indicates the presence of a single amplicon at the end of the PCR reaction. If multiple peaks were detected, it would indicate that multiple PCR products were produced and the PCR assay would require further optimization and development. Comparison of mRNA expression levels between species or other circumstances, by qPCR, may be achieved through the addition of a synthetic RNA to correct for differences in RNA isolation and reverse transcription. The limitations of Real-time PCR are the increased costs, the specialized thermocyclers, and the limitation of precision in quantifying the starting quantity of target NA sequences. RT-PCR can be accomplished through commercial kits such as Cells-to-CT 1-Step TaqMan Kit from Thermo Fisher [23], Qiagen OneStep Ahead RT–PCR [24] or Integrated DNA Technologies PrimeTime qPCR assay kits [25]. Human ACTB or mouse Actb gene is often as the control [25, 26]. Zeng Q et al, for example, normalized RT-PCR results to the housekeeping genes ACTB or GAPDH for human cells and Rpl19 or Gapdh for mouse cells [27]. Corman VM et al quickly established a real-time PCR assay for detecting 2019-nCoV [28].
PCR arrays utilize Real-time PCR thermocyclers are based on the Real-time PCR SYBR green assay. PCR arrays have multiple primer sets within a 96-well plate to measure the expression of up to 88 genes and eight normalization or control reactions for a given sample. PCR arrays measure a single gene-specific product within each well of the plate. These arrays normally include control reactions and allow quantitative gene expression results. Arrays are often generated to analyze the expression of a group of genes involved with a specific biological pathway, such as DNA repair, cell cycle, or oxidative stress. The advantage to PCR arrays is that expression of many different genes can be obtained at once for a sample. The limitation is that each sample must be processed separately, so it is time-consuming and also costly. De Cecco M et al employed the Qiagen RT2 Profiler Human Type I Interferon Response PCR Array (PAHS-016ZE-4) to study the involvement of L1 retrotransposon during cellular senescence [29]. D Thomas et al examined human ECM and adhesion molecules through RT2 profiler PCR array (PAHS-013Z) from QIAGEN / SABiosciences [30].
Microfluidic chip PCR utilizes microfabrication and microfluidics to amplify DNA much more quickly than conventional or Real-time PCR. Besides, the small size and integration with detection components offer improved portability and field accessibility to PCR. Digital and drop digital PCR partitions NA molecules and quantifies end-point PCR product without the need for a standard curve. This allows for very precise copy number determination and provides detection for very low copy number NA sequence targets. Although Chip PCR is robust and precise, its accessibility to many researchers is fairly low. This is due to the need for microfabrication equipment, or purchasing premade chips, which can be quite costly. However, Chip PCR is evolving rapidly, and the costs will likely be lowered significantly soon.
Many other PCR methods are well utilized in biological research. Colony PCR can be used to screen for the presence of a specific genomic insert from bacterial colonies without the need for culturing or plasmid purification [31]. Genotyping often uses allele-specific PCR [32]. Epigenetic research is heavily reliant upon methylation-specific PCR. This technique detects the methylation of CpG islands in genomic DNA [33]. Touchdown PCR reduces non-specific amplification by systematically lowering the annealing temperature as a reaction progresses. This technique can improve the yield of a specific target sequence [34]. Nested PCR can increase the sensitivity of detection. For example, de Goffau MC et al used a nested PCR-qPCR approach to detect the Streptococcus agalactiae sip gene in human placental samples [35].
Although PCR is a ubiquitous method, opportunities to reduce the length of time required to obtain results and to reduce the reaction volumes are abundant. The speed of a reaction is dependent on the types of heating and cycling mechanisms as well as reaction volume, and thus these are prime targets for assay enhancement. Metal-block-based PCR systems, which use indirect conductive heating, are widely utilized, yet the block has a high thermal mass and requires relatively large reaction volumes, making the rates of thermal change fairly slow (3ºC/s) [36]. The Roche Lightcycler uses convective heating and requires just several microliters of volume, so the speed of temperature change is much quicker (10ºC/s) [37]. Microchip PCR devices offer even greater speed and volume reduction. These are most frequently made from silicon or glass, which are thermally conductive. These chips are heated by thermoelectric heating, convection-based rotary platforms, or embedded resistive heaters [38-40] and require just a few microliters of volume. Direct heating from infrared radiation via a tungsten filament lamp has been used in microchip PCR [41]. IR mediated direct heating can also be carried out by lasers [42, 43]. Both give PCR results in minutes and require just picoliters of volume. See Labome review on PCR machines for a comprehensive review.
False positive and negative PCR results are detrimental to the usefulness of a PCR assay, and so optimization to prevent these is imperative. False positive results occur when DNA amplification is detected even though no starting temple NA was added to the reaction. This occurs when contamination is present. Clean laboratory practices are necessary to avoid aerosolization of DNA so that it isn’t unintentionally introduced into reactions. False negative results occur when no NA amplification is detected, yet the target NA is present. This can be caused by poor primers, suboptimal thermocycling parameters, or quantities of product amplicon that are below the limit of detection of a system. To reduce the occurrence of false negatives, the optimization of PCR assay design and cycling conditions is necessary. This includes the selection of quality primers [44-46], optimization of temperatures during cycling, and the use of quality template NA.
Real-time PCR offers significant advantages over conventional endpoint PCR. Real-time PCR utilizes detection systems that measure fluorescence indicative of DNA amplification within a closed tube, normally in a 96 well plate format. This negates the need for post-PCR manipulations or detection via agarose gel electrophoresis, and thus significantly reduces the time required to obtain results. The detection of the PCR product occurs after every cycle, and thus allows for the measurement of reaction kinetics. The fluorescent detection of PCR product is also much more sensitive than detection of DNA via agarose gel electrophoresis, and so this technology can identify as little as a two-fold difference in DNA quantities, something that is not possible with conventional PCR and agarose gels. Furthermore, when utilizing probe based Real-time assays, detection of the PCR amplicon is sequence specific.

DNA binding dyes: Real-time PCR employs fluorescent dyes or probes that interact with the PCR products. The two primary types of fluorescent detection are DNA binding dyes, such as SYBR Green, or fluorescently tagged sequence-specific probes, such as TaqMan or Molecular Beacon probes. When the DNA binding dyes attach to any double-stranded DNA segment, they emit a fluorescent signal [47]. When in the presence of single-stranded nucleic acids, these dyes do not attach to the NA and emit only low levels of fluorescence. Although SYBR Green is commonly used, several other DNA binding dyes are also utilized. These include SYTO 9 [48], SYTO-13, SYTO-82, [49] and EvaGreen [50, 51]. Since detection of a fluorescent signal from these dyes is not sequence-specific, melting temperature analysis must be performed to ensure the production of a single PCR product [52]. See Figure 4 for an example of a single PCR product in a melt curve plot. If a melt curve had multiple peaks, it would indicate the presence of multiple PCR products, and the PCR assay would require further optimization. Although this may be time-consuming, SYBR Green and other DNA binding dyes remain popular because they are significantly less expensive than most alternative probe-based assays.
Nuclease dependent probes: Sequence-specific fluorescently labeled probes are the second main type of detection chemistry utilized in Real-time PCR. These probe systems can be nuclease dependent or simply hybridization probes. The nuclease cleaved probes include TaqMan, HybProbe (two oligonucleotides), minor groove binding (MGB), and locked nucleic acid (LNA) probes. These probes are complementary to a target nucleic acid sequence within the PCR amplicon, and they have both a reporter and a quencher fluorophore covalently attached to opposing ends. These systems utilize Florescent Resonance Energy Transfer (FRET) technology. When the dyes remain near one another, the fluorescent signal of the reporter dye is quenched, preventing any detectable signal. When the dyes are separated, the reporter dye’s fluorescence is unquenched and thus detectable [53]. A probe anneals to a sequence internal to the PCR primers’ binding sites, and as the Taq DNA Polymerase enzyme extends the primers to produce the PCR product, its 5’ exonuclease activity cleaves the end of the probe. The cleavage removes the quencher dye and allows excitation of the reporter dye, resulting in a fluorescent signal. Cycling probe technology (CPT) probes differ from the TaqMan type probes in that they include an RNA nucleotide. These probes form an RNA-DNA duplex upon hybridization to the target sequence. Then RNase H enzyme is used to cleave the quencher dye from the probe [54].
Hybridization probes: Although hybridization probes are also sequence-specific, they do not require the exonuclease cleavage of a dye from the probe. These include Molecular Beacons, which have a loop region between two inverted repeats, creating a hairpin structure. When the probe is denatured and anneals to a target sequence, the hairpin is released, and the fluorophores are separated from one another enough that the reporter dye generates increased fluorescence [55]. Other detection probe technologies include the Scorpion (probe and one PCR primer are combined in one molecule [56] ), and LUX (Light Upon eXtension) assays [57]. The Lux primer probe has a dye near its 3’ end that is quenched by the hairpin structure of the primer. Once it binds to a target sequence and DNA Polymerase extends the sequence, the dye’s signal increases [57].
Real-time PCR thermocycler systems detect fluorescence, which is proportional to the accumulation of PCR amplicon. However, the analysis is performed only with the data from the exponential phase of the reaction, because this is the optimal point for precise quantitation. The threshold cycle (CT) is the cycle number at which the fluorescence within a reaction crosses the threshold, a level of signal set above the baseline but within the exponential phase of the amplification. Two general methods of quantification determination are used for Real-time PCR. Relative quantification uses a relative expression ratio of the amount of target sequence from control versus experimental treatments, as normalized to the expression of a control gene such as Gapdh, Hsp90ab1, and Gusb [29] or cyclophilin A [58]. It is critical to use a control gene that has uniform expression across treatments. For instance, GAPDH served as the normalization control in experiments with human cells and the mean of Gapdh, Hsp90ab1, and Gusb served as a normalization control of RT–qPCR experiments with mouse tissues (with the exception of liver) [29]. The primary advantage to relative quantification is the reduced assay development time because the creation of a calibration curve is not required. Both a relative standard curve method and the comparative CT method (ΔΔCT) are commonly used. A mathematical model to calculate the later was described by Pfaffl [59].
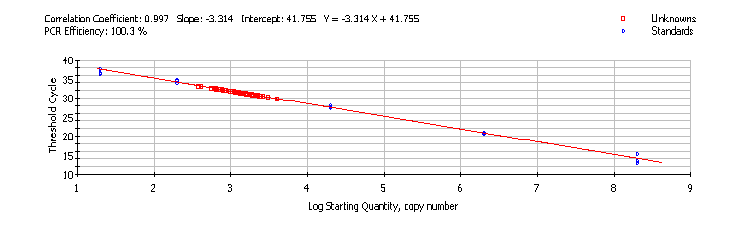
Absolute quantification measures the actual amount of starting target sequence in a sample by comparing the sample amplification signal to that of a standard curve of target DNA. Calibration curves may be based on dilutions of nucleic acid molecules such as recombinant plasmid DNA containing a subcloned amplicon, Real-time PCR product, or large synthesized oligonucleotides. Plasmid DNA is widely used because it has higher stability than PCR products or oligos. However, the shorter templates are less time consuming to prepare. A plasmid standard curve is generated by performing Real-time PCR on known concentrations of the plasmid (with amplicon insert) and graphing the CT values against the log of the initial target copy number (Figure 5). The CT value is inversely proportional to the log of the initial copy number [60]. Initial copy numbers of target sequences for experimental samples are determined by linear regression against the standard curve [61].
Since the 1990s, innovative miniaturization of PCR reactions and thermocyclers has been well pursued in the form of microfluidic chip PCR. For instance, in 1998, Northrup et al reported their construction of a miniature analytical thermal cycling instrument (MATCI) that was silicon-based and contained chambers with integrated heaters [62]. The device performed fluorescence measures in real-time as PCR cycling occurred, and was as portable as a briefcase. Since then, the field of microfluidic chip PCR has exploded, and advancement of the technology has included multiple fabrication materials and architectures [63].
A majority of PCR chip reaction wells are silicon-based. Silicon has excellent thermal conductivity, which allows for quick temperature changes during thermocycling. However, chips have also been fabricated from glass, polymers including polycarbonate and polydimethylsiloxane, ceramic, and 317 stainless steel. Chip architectures are either stationary chamber based or dynamic continuous flow based. The stationary chamber chips have a PCR reaction solution that is held stationary while the temperature of the chamber is cycled. These chips may be single or multi-chambered. The dynamic continuous flow based chips have amplification while the sample is pumped through a microfluidic channel during temperature cycling. With these chips, the flow rate of the sample determines the time for temperature transitions. Microchip PCR offers the benefit of short assay times, low consumption of reagents, rapid rates of heating and cooling, and reduced power consumption. Microchip PCR may require downstream handling for DNA detection, such as agarose gel or capillary electrophoresis. However, capillary electrophoresis, as well as fluorescence detection or DNA hybridization methods of DNA detection, may be included on the chip [38, 64]. Microchip PCR continues to be developed with advanced integration of on-chip detection systems and optimization of both fabrication materials and temperature cycling methodologies.
The microfluidic chamber based digital PCR (cdPCR) method is capable of absolute quantification of nucleic acids without the use of standard curves [65]. With digital cdPCR, the sample is partitioned into discrete water-in-oil droplets that may have no target NA or may have one or more copies. The presence of the target sequence is measured at the end of PCR amplification, and the concentration is calculated from the fraction of positive partitions versus the total number of partitions. The “Drop” digital PCR (ddPCR) is a cutting-edge version of digital PCR. It has twenty-five times more partitioned droplets, which allows for very accurate copy number estimates [66]. Q Xu et al used QX200 AutoDG Droplet Digital PCR System (Bio-Rad) and the SARS-CoV-2 Droplet Digital PCR kit (Bio-Rad) to detect and quantify SARS-CoV-2 RNA in FFPE tonsil and adenoid tissues [67]. R Garcia-Martin et al used ddPCR to obtain miRNA copy number from exosomes and cells [68]. M Aubert et al quantified the presence of latent herpes simplex virus genpme in mouse cells with ddPCR [69]. de Morree A et al compared the expression of Pax3 RNA isoforms between diaphragm and limb muscle stem cells through digital PCR [70]. Lee J et al measured allele-specific expression of LMNA wild-type and mutant allele in iPSC-derived cardiomyocytes with ddPCR [71]. Researchers have directly compared ddPCR and real-time PCR platforms for the detection of low abundance DNA and found that ddPCR was superior over real-time PCR [72]. In February 2019, Bio-Rad released the first FDA-cleared droplet digital PCR system and BCR-ABL fusion test for monitoring chronic myeloid leukemia treatment response.
- Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol. 1986;51 Pt 1:263-73 pubmed
- Mullis K, Faloona F. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 1987;155:335-50 pubmed
- Saiki R, Scharf S, Faloona F, Mullis K, Horn G, Erlich H, et al. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science. 1985;230:1350-4 pubmed
- Malorny B, Tassios P, Radstrom P, Cook N, Wagner M, Hoorfar J. Standardization of diagnostic PCR for the detection of foodborne pathogens. Int J Food Microbiol. 2003;83:39-48 pubmed
- McBride L, Koepf S, Gibbs R, Salser W, Mayrand P, Hunkapiller M, et al. Automated DNA sequencing methods involving polymerase chain reaction. Clin Chem. 1989;35:2196-201 pubmed
- Decorte R, Cassiman J. Forensic medicine and the polymerase chain reaction technique. J Med Genet. 1993;30:625-33 pubmed
- Schlenk J, Seidl S, Braunschweiger G, Betz P, Lederer T. Development of a 13-locus PCR multiplex system for paternity testing. Int J Legal Med. 2004;118:55-61 pubmed
- Saiki R, Bugawan T, Horn G, Mullis K, Erlich H. Analysis of enzymatically amplified beta-globin and HLA-DQ alpha DNA with allele-specific oligonucleotide probes. Nature. 1986;324:163-6 pubmed
- Herman J, Graff J, Myöhänen S, Nelkin B, Baylin S. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821-6 pubmed
- Don R, Cox P, Wainwright B, Baker K, Mattick J. 'Touchdown' PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res. 1991;19:4008 pubmed
- Wittwer C, Ririe K, Andrew R, David D, Gundry R, Balis U. The LightCycler: a microvolume multisample fluorimeter with rapid temperature control. Biotechniques. 1997;22:176-81 pubmed
- Khandurina J, McKnight T, Jacobson S, Waters L, Foote R, Ramsey J. Integrated system for rapid PCR-based DNA analysis in microfluidic devices. Anal Chem. 2000;72:2995-3000 pubmed
- Liao C, Lee G, Liu H, Hsieh T, Luo C. Miniature RT-PCR system for diagnosis of RNA-based viruses. Nucleic Acids Res. 2005;33:e156 pubmed
- Huhmer A, Landers J. Noncontact infrared-mediated thermocycling for effective polymerase chain reaction amplification of DNA in nanoliter volumes. Anal Chem. 2000;72:5507-12 pubmed
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365-86 pubmed
- Schneeberger C, Speiser P, Kury F, Zeillinger R. Quantitative detection of reverse transcriptase-PCR products by means of a novel and sensitive DNA stain. PCR Methods Appl. 1995;4:234-8 pubmed
- Monis P, Giglio S, Saint C. Comparison of SYTO9 and SYBR Green I for real-time polymerase chain reaction and investigation of the effect of dye concentration on amplification and DNA melting curve analysis. Anal Biochem. 2005;340:24-34 pubmed
- Gudnason H, Dufva M, Bang D, Wolff A. Comparison of multiple DNA dyes for real-time PCR: effects of dye concentration and sequence composition on DNA amplification and melting temperature. Nucleic Acids Res. 2007;35:e127 pubmed
- Ririe K, Rasmussen R, Wittwer C. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem. 1997;245:154-60 pubmed
- Holland P, Abramson R, Watson R, Gelfand D. Detection of specific polymerase chain reaction product by utilizing the 5'----3' exonuclease activity of Thermus aquaticus DNA polymerase. Proc Natl Acad Sci U S A. 1991;88:7276-80 pubmed
- Duck P, Alvarado Urbina G, Burdick B, Collier B. Probe amplifier system based on chimeric cycling oligonucleotides. Biotechniques. 1990;9:142-8 pubmed
- Tyagi S, Kramer F. Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol. 1996;14:303-8 pubmed
- Whitcombe D, Theaker J, Guy S, Brown T, Little S. Detection of PCR products using self-probing amplicons and fluorescence. Nat Biotechnol. 1999;17:804-7 pubmed
- Nazarenko I, Lowe B, Darfler M, Ikonomi P, Schuster D, Rashtchian A. Multiplex quantitative PCR using self-quenched primers labeled with a single fluorophore. Nucleic Acids Res. 2002;30:e37 pubmed
- Pfaffl M. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45 pubmed
- Higuchi R, Fockler C, Dollinger G, Watson R. Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology (N Y). 1993;11:1026-30 pubmed
- Bustin S. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol. 2000;25:169-93 pubmed
- Northrup M, Benett B, Hadley D, Landre P, Lehew S, Richards J, et al. A miniature analytical instrument for nucleic acids based on micromachined silicon reaction chambers. Anal Chem. 1998;70:918-22 pubmed
- Zhang C, Xing D. Miniaturized PCR chips for nucleic acid amplification and analysis: latest advances and future trends. Nucleic Acids Res. 2007;35:4223-37 pubmed
- Vogelstein B, Kinzler K. Digital PCR. Proc Natl Acad Sci U S A. 1999;96:9236-41 pubmed
- Materials and Methods [ISSN : 2329-5139] is a unique online journal with regularly updated review articles on laboratory materials and methods. If you are interested in contributing a manuscript or suggesting a topic, please leave us feedback.