An overview of research methodology in worms.
In 1963, when Sydney Brenner (who died on April 5, 2019 at the age of 92) was exploring the next new challenge in biology, he concluded that studying the organizing principles of multicellular organisms, in particular, the organization of the nervous system and embryonic development, was the most exciting area of study for the future [8]. However, he felt that a new experimental system must be found to unravel the complexities of multicellular systems. And so, Caenorhabditis elegans as a model system was born. Sydney Brenner published his pioneering research in 1974 on the isolation of mutants that affect various aspects of the nervous system [9]. Since then, his strategy has been adopted and modified by researchers worldwide to address fundamentally important questions in all aspects of biology.
C. elegans is small, transparent, free-living, soil nematode. C. elegans or ‘worms’ as they are famously known as, are easy to grow, can be maintained in large numbers, have a short life cycle, and can be frozen for long periods of time. C. elegans has six chromosomes: five pairs of autosomes (I, II, III, IV, and V) and the X chromosome. The hermaphrodites have two X chromosomes (designated as XX), and the males have one X chromosome (designated as XO). The hermaphrodites are easily identified by the presence of the vulva and the males by their fan-like tail. C. elegans has a life span of about 14 days. The reproductive life span of the worm varies with the temperature at which they are maintained. The worms will go from an egg to an egg-laying parent in 5.5 days at 15°C, 3.5 days at 20°C and 2.5 days at 25°C. The hermaphrodites can self-fertilize or mate with males. The male worms are naturally rare and arise spontaneously as a result of non-disjunction in the hermaphrodite germline with a frequency of 0.1%. Following self-fertilization, the hermaphrodite produces about 300 genetically identical progeny. However, when hermaphrodites mate with males, 50% of the progeny are males, and the progeny number rises to about 1200 to 1400. No wonder, the worms can be easily maintained in large numbers! The males are great for isolation and maintenance of mutations and moving them between strains.
Following fertilization, an egg undergoes a series of cell divisions and is then laid a few hours later. After hatching, the worms pass through four larval stages (L1-L4), each of which ends in a molt. Unfavorable conditions, such as scarcity of food, high temperature or high population density trigger the L2 larvae to enter an alternative developmental program called the dauer stage. Interestingly, the dauers can survive without food for months. When conditions become favorable again, they resume development to reach adulthood.
The worms follow an invariant pattern of development, which allows researchers to track biological events at the level of single cells. As worms are transparent throughout their life cycle, they can be viewed at the cellular level in live preparations by differential interference contrast (DIC) microscopy. The Green Fluorescent Protein (GFP)-tagged reporters can be easily injected into worms, and offer the advantage of making anatomical observations, analysis of expression pattern and protein localization in live worms. Above all, C. elegans is a powerful system for investigating gene function through genetic screens. The ease of generating mutants, being able to examine these mutations in a homozygous state and being able to preserve them as frozen stocks make worms a very attractive system for genetic analysis. The worms have many tissues and organs that are present in higher organisms, such as the nervous system, digestive tract, muscles, and reproductive system. Even though all these systems are present in the worm in a simplified state, the basic molecular architecture is similar to that of higher organisms. There are less molecular interactions to navigate through, but the roles of the major players have been remarkably conserved during evolution. The worms also exhibit complex behaviors that are modulated by their environment as well as experience [10]. Since the complete sequence of the worm genome became available, C. elegans homologues have been identified for a large number of human genes, and many signal transduction pathways have been found to be conserved between C. elegans and humans [11]. Therefore, C. elegans has been successfully used to address fundamentally important questions in many fields, including full-length transcriptome [12], germline gene expression [13], chromosome structure during embryonic development [14], ageing [15, 16], neurobiology [17], mating behavior, and even rudimentary forms of learning, and more recently, for understanding microbial pathogenesis and innate immunity [18]. Cook SJ et al published the whole-animal connectomes of C. elegans [19]. Packer JS et al identified 502 terminal and pre-terminal cell types and resolved the lineage of individual cells during its embryogenesis with single-cell RNA-seq [20].
In this article, we discuss some of the important methods used in C. elegans research. The following sections describe popular methods for transforming worms, making transgenics, generating knockouts and knockdowns, and optical and electrophysiological methods for studying worm behavior.
Mutations provide an entry point into the myriad and intricate world of biological pathways. As a recent example, CY Shi et al used Caenorhabditis elegans with a null mutation in ebax-1 to investigate the role of ZSWIM8 in microRNA stability [21]. Mutations can be generated either through genetic screens or by targeted deletion strategies. The C. elegans Genetic Center (CGC) maintains a repository of worm mutants, which are available to all academic researchers for a small fee. In 1998, C. elegans became the first multicellular organism to have a completely sequenced genome, and the wealth of information that was generated by this endeavor made genetic analysis even more amenable and more sophisticated. Therefore, genetic screens have been used to explore practically every aspect of worm biology. In recent years, RNA interference (RNAi) has become a very important and a powerful tool for silencing genes and for conducting genome-wide screens, so have CRISPR-based systems [1, 22].
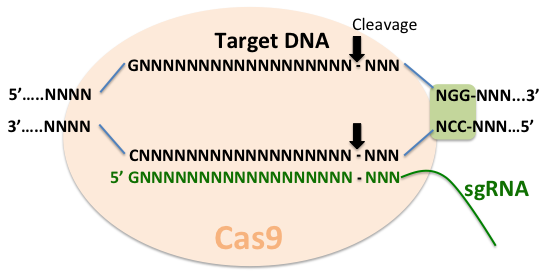
Targeted changes in the genome offer the ability to address fundamentally important questions. As discussed in the previous section, ways to manipulate gene function, such as RNAi, offered a convenient way to carry out high throughput genetic screens. However, RNAi leads to only temporary inhibition of gene function and sometimes, unpredictable and undesirable off-target effects. A recent adaptation of the bacterial adaptive immune system - clustered, regularly interspaced, short palindromic repeats (CRISPR) and CRISPR-associated (Cas) systems, offers important advantages over existing tools for studying gene function. As a part of the defense strategy in bacteria, these systems incorporate foreign DNA into the host CRISP loci to generate short CRISPR RNAs (crRNA), which then serve as guides for the effector endonucleases, such as Cas9, to target invading DNA based on sequence complementarity.
The CRISPR-Cas9 system has been applied in many model organisms, including C. elegans [1, 22]. In order for it to work, the system requires the presence of NGG motif immediately downstream of the target sequence and a separate trans-acting RNA (tracrRNA or trRNA) that is partially complementary to the crRNA. The simplified system combines the crRNA and trRNA into a single synthetic guide RNA (sgRNA) [23]. Target specificity is achieved by base pairing between the 5’ end of the sgRNA and the target DNA. Therefore, a very large repertoire of substrates can be accomplished by changing the 5’ end of the sgRNA. The Cas9-induced breaks are repaired by homologous recombination. By providing engineered homologous repair templates, insertions, deletions, point mutations and GFP insertions can be achieved [1].
Ethyl methane sulfonate (EMS) is a commonly used reagent for inducing mutations [8]. The mutations recovered with EMS are usually G/C-A/T transitions and occasionally, small deletions [24]. Mutagenesis is performed with healthy, young adult worms. Following mutagenesis, the worms are placed on petri plates to produce homozygous progeny, which are examined for the phenotype of interest. The mutant worms are analyzed in detail to determine the pathway in which the genes function. A saturated screen for a phenotype may uncover many or all of the components in a particular pathway. However, sometimes, secondary modifier screens, which look for second site mutations that either suppress or enhance the effect of the original mutation, are necessary to identify additional components of the same pathway [8]. It is important to note that some phenotypes may not be observed in the first generation and the mutant worms may have to be maintained for many generations for the phenotype to become obvious. Some phenotypes may not be easily observed under the dissecting microscope and specific fluorescent markers may be used to report on the phenotype [8].
The most commonly used chemical mutagenesis strategies rarely lead to complete deletion of genes, in fact, the majority of the deletions generated by this method are small and may not be functional nulls. Chemical mutagenesis invariably leads to background mutations that must be out-crossed. In addition, mapping of mutant genes with even the most sophisticated methods can sometimes be tedious and expensive. In 2010, Erik Jorgensen’s group reported a way of making targeted deletions in the worm genome using the Drosophila Mos1 transposon [2]. Transposons are mobile DNA elements that can jump between chromosomes. For generating a Mos-mediated deletion, an injection strain is generated by selecting the gene of interest near the Mos1 transposon and crossing it into the unc-119 mutant background. The injection mix contains three plasmids: (i) MosI transposase for excision of the transposon, (ii) the unc-119 wild type gene flanked by DNA sequence homologous to the region where the strand break occurs but lacking the gene of interest, for selection and for strand repair, and (iii) a red fluorescent reporter to mark the extrachromosomal array. The Mos1 transposase excises out the transposon in the vicinity of the gene of interest and leaves a chromosomal break. The DNA break is repaired by using the second injected plasmid as a template, which results in the deletion of the gene of interest in the worm progeny. During this process, the unc-119 wild type gene is also inserted, which serves as a selection marker. Following injection and recovery, the worms are allowed to starve. As the unc-119 mutants cannot form dauers, only the rescued worms survive. The rescued worms without the red fluorescent reporter are ones that are deleted for the gene of interest. It has been determined that the maximum number of DNA base pairs that can be reliably deleted by this method is 25,000 [2]. The worm genes vary anywhere from 700 base pairs to 30,000 base pairs, therefore, this technique can be applied to precisely and completely knockout most genes in C. elegans. Currently, 14,000 molecularly mapped Mos1 insertions are available from the NemaGENETAG consortium, providing a valuable resource for generating Mos1-based deletion strains. A variation of this approach is mos1-mediated Single Copy Insertion (MosSCI), which can introduce a transgene into a chromosome, even in the germline. Cabianca DS et al, for example, introduced MRG-1–mCherry and others into ttTi5605 on Chr II [25].
In addition, Lightfoot JW et al integrated the self-1 cDNA sequence from P. pacificus into the genome of C. elegans strain HT1593 containing a mutation in unc-119 through injection of an appropriate plasmid mix to study the self recognition in predatory nematodes [26].

RNA interference (RNAi) is a biological process for inhibiting gene expression by causing the destruction of specific mRNA molecules. Since the groundbreaking discovery of RNAi in C. elegans by Craig Mello and Andrew Fire, RNAi has become a very popular research tool for studying gene function [27, 28].
In C. elegans, RNAi has been applied to systematically shut down the expression of each gene to conduct genome-wide screens. For RNAi, double-stranded RNA (dsRNA) can be introduced in the worms by injecting, soaking or feeding. For example, Lardennois A et al injected or fed double-stranded RNAs against spc-1, pak-1, fhod-1 and unc-112 to investigate the role of mechanical forces in body-axis elongation [29]. In worms, RNAi ‘spreads’ to produce a systemic response following a localized injection, therefore, dsRNA can be injected into the intestine, gonad or the body cavity. Although the results are usually equivalent regardless of the site of injection, some RNAi phenotypes may be stronger after a gonad injection. The injected worms must be examined at different time points to determine the optimum screening time. Microinjections are not practical for performing large-scale RNAi screens. Soaking the worms in a concentrated solution of dsRNA also induces RNAi as dsRNA is ingested through the pharynx [30]. The soaking approach is not as tedious as microinjections and also amenable to high throughput screening, however, it is expensive due to the large amount of in vitro transcribed dsRNA required for soaking the worms.
Unlike microinjections and soaking, RNAi screens by feeding are far less labor-intensive and also inexpensive and are commonly used [16]. For example, Lardennois A et al conducted a RNAi screen with 356 essential genes from the Ahringer RNAi library in the pak-1 mutant and identified alpha-spectrin gene spc-1 as a strong genetic enhancer of pak-1 [29]. With a good RNAi library, most of the genome can be screened in a relatively short period of time. The RNAi library is a collection of bacterial strains, which produce the dsRNA corresponding to different worm genes upon induction. The developmental stage at which the dsRNA is introduced into the worms depends largely on the phenotypes to be scored. Regardless of the stage at which dsRNA is fed to the worms, for most screens, researchers start with a synchronized population of worms, which can be obtained by standard bleaching protocols. Synchronization ensures that all the worms being screened are at the same developmental stage. RNAi by feeding can be performed on petri plates or in liquid culture. RNAi by feeding in liquid culture is ideal for scoring easily observable phenotypes such as sterility, lethality, slow growth and dramatic changes in GFP expression. Plate feeding is more suitable for observing subtle phenotypes. Certain phenotypes, such as changes in movement, feeding or behavior can be examined only on plates.
Certain factors must be kept in mind while interpreting the results of RNAi screens. It has been observed that different genes have different sensitivities to RNAi. As RNAi knocks down gene expression by causing the degradation of the messenger, any residual protein inherited from the mother may be enough to mask the phenotype. The phenotype may also be masked in the case of genes that encode for proteins with long half-lives. Antibodies may be used to determine if the protein levels have been significantly reduced following RNAi. Also, different genes that share significant sequence similarity may be knocked down by the same dsRNA [31]. Interestingly, almost all mRNAs in the worm nervous system are refractory to RNAi. Therefore, RNAi screens for neuronal genes may be conducted in the RNAi supersensitive eri-1 mutants [32] or sid-1(pk3321) mutants [17]. The eri-1 gene encodes for a negative regulator of RNAi [32]. The sid-1 gene, a dsRNA transporter, is required for RNAi, and is re-expressed in neurons via a punc-119:sid-1 transgene in sid-1(pk3321) mutants, thus rendering the mutants not deficient in RNAi in neurons only.
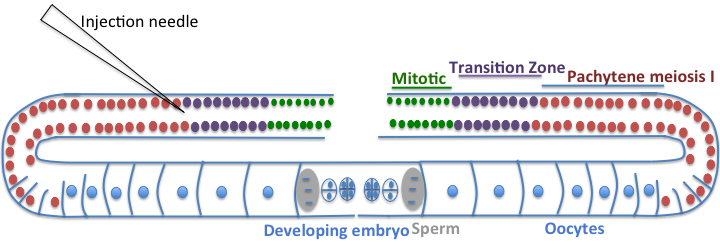
Microinjection is one of the most common and quickest ways of introducing DNA into worms and transforming the germline [33]. The technique has been widely applied for studying genes of interest, rescuing mutant phenotypes for cloning, introducing genes tagged with reporters to analyze spatial and temporal variations in expression pattern, and studying regulatory elements [34]. For microinjection, one needs healthy, young adult hermaphrodites. The DNA is injected in the gonad. The worm gonad is comprised of two symmetrical U-shaped tubes [35]. Each tube is organized in a distal to proximal manner, with the proximal end being the one that is closest to the vulva. The distal end of the germline on each side is a syncytium of nuclei surrounding a central cytoplasmic core. These cells are mitotic and undifferentiated. As these cells enter the bend of the gonad (oviduct), they are surrounded by plasma membrane to form oocytes. The oocytes undergo meiosis as they pass into the proximal part of the gonad tube, and are fertilized by hermaphrodite-derived sperm or by male-derived sperm that is introduced during copulation. The microinjection is done in the cytoplasm of the syncytial gonad, which ensures that the injected DNA is introduced into the nuclei of oocytes and consequently into fertilized eggs [36]. To determine if the transformation has been successful, an easily selectable marker is co-transformed. The co-injected marker should allow easy identification of the transformed worms but should not interfere with the expression of the DNA being tested. A commonly used marker is the rol-6 mutant gene [35]. This gene encodes for mutant cuticle collagen and produces a dominant ‘roller’ phenotype, which is easily spotted under a dissecting microscope. Though it is easy to spot, it has certain disadvantages; the roller phenotype is suppressed in some mutant backgrounds, the roller males have reduced fertility and make genetic crosses difficult, in addition, the twisted cuticle of these worms makes an analysis of live worms difficult. An alternative to the use of rol-6 mutant is the GFP tagged reporter as a co-injection marker. The GFP reporters have become quite popular as co-injection markers, but their use requires a fluorescent dissecting microscope [37]. Zullo JM et al, for example, used Prab-3::mCherry from Addgene ( 19359) and Pmyo-2::mCherry from Addgene ( 19327) as coinjection markers [17].
The injected DNA molecules assemble into large extrachromosomal arrays or concatemers that contain many copies of DNA [38]. The transmission of the arrays is dependent on size; arrays greater than 700 kb are transmitted through the germline as extrachromosomal elements, whereas smaller arrays are lost unless integrated into the genome [39]. Genomic integration of the extrachromosomal arrays is rare but it does occur at a low frequency. There has been no systematic analysis of the correlation between the copy number of the injected DNA and mRNA levels or protein expression levels [36]. However, it has been observed that though the structure of the arrays remains rather stable, the expression levels from repetitive arrays may change over time, and the expression level in the siblings may vary. In addition, the expression pattern of the transgene may not reflect that of the endogenous gene due to overexpression or silencing.
The arrays, in general, tend to be unstable because of their repetitive structure and may be lost over time. Repetitive arrays are strongly silenced in the germ cells, and this problem may be circumvented by the use of ‘complex’ arrays [40]. The complex arrays can be formed by including fragmented yeast genomic DNA in the injection mix [37]. It is thought that silencing is prevented because the genomic DNA fragments are incorporated and limit the formation of tandem arrays. However, it has been observed that even complex arrays may be silenced in the germline after a few generations [33].
The extrachromosomal arrays may be integrated into the genome to prevent problems arising from their inherent genetic instability. The transgenic strains are subject to gamma or UV irradiation to integrate the arrays, however, this invariably leads to mutations in the parent strain that must be out-crossed by mating with wild type males. Despite the problems associated with microinjections, it remains the quickest way of generating transgenics, and well-behaved and useful strains are routinely generated using this technique.
As mentioned in the preceding section, the generation of transgenics for genes that are either dose sensitive or are silenced when present in high copy numbers has been especially tedious [41]. Microparticle bombardment can produce integrative transformants, which can overcome many of the problems that are associated with the presence of extrachromosomal arrays in high copy numbers. Zullo JM et al, for example, generated the SPR-4::GFP reporter line trhough microparticle bombardment [17]. As is the case with the transformation of bacteria and yeast, this technique relies on transforming a population of worms instead of individual worms [33]. The gene of interest is cloned on the same plasmid that carries the selection marker [42]. A small amount of DNA is coated on gold particles, accelerated to high speeds and allowed to penetrate the worms using the biolistic bombardment apparatus. It is thought that low copy integrants arise from the entry of the coated gold particles into the oocyte nuclei. The selection of integrants by bombardment is based on the rescue of unc-119 mutant worms by the wild type unc-119 gene, which is present on the same plasmid as the gene of interest. The unc-119 mutants are viable but unable to form dauers. Following transformation, the worms are allowed to recover, feed on bacteria, reproduce and starve before the screening is performed [40]. Under these conditions, only the rescued worms survive, which are analyzed further for the presence of the gene of interest.
The generation of transformants by microinjection takes 7-10 days, whereas it takes 3-4 weeks by bombardment. However, stable integrants can be obtained from bombardment without any further processing. On the downside, microparticle bombardment is more labor-intensive, expensive to set up and requires continuing investment for the supplies.
The ability to regulate transgene expression with binary systems has been a very important development in cell biology. In a binary system, one transgene contains a specific promoter, which drives the expression of an exogenous transcription factor while the other transgene carries the promoter that is activated only by the exogenous transcription factor. Therefore, the expression pattern as well as the expression level of the gene of interest is regulated only by that particular transcription factor. The transgenes are brought together by a genetic cross to induce expression of the gene of interest. The GAL4-UAS binary system has been used with unparalleled success in Drosophila, however, it could not be adapted to worms. Liqun Luo’s research group recently reported a new repressible binary system, called the Q-system for use in Drosophila and cultured cells [4]. The system has been successfully adapted for use in C. elegans [5]. The Q system is based on regulatory genes from the Neurospora crassa qa gene cluster. The transcriptional activator, QF binds to 16 bp QUAS sequences to activate transcription of target genes under the control of QUAS sites [4, 43, 44]. The transcriptional repressor, QS binds to and inhibits QF to suppress the activation of target genes [44]. A small molecule, quinic acid can bind to QS, which releases QF to induce gene expression [4, 5, 43]. To test whether the Q system works efficiently in worms, the QF construct and the QUAS::GFP construct were maintained separately and then brought together by crossing [5]. Robust GFP expression was observed only when both transgenes were present in the worms. As microinjections yield high copy number extrachromosomal arrays that are subject to silencing, the components of the Q system can be integrated into the genome.
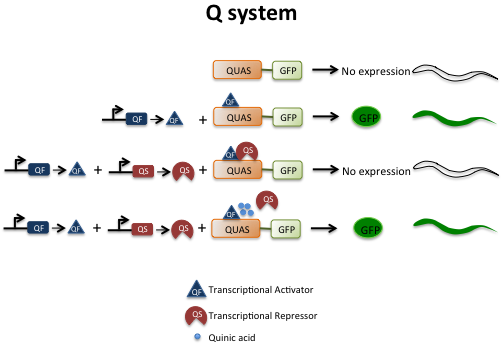
The binary system offers a big advantage over the conventional promoter-driven fusions; it is difficult to modulate expression from promoter-driven fusions, whereas in the Q system, expression can be suppressed using QS or turned on using quinic acid. Thus, it is possible to turn genes on or off at specific times. The libraries of QF drivers and QUAS effectors can be established that can be combined by genetic crosses. By turning off or turning on the expression of genes, these libraries can be used to systematically address the temporal and spatial effects of genes in different biological processes.
The neural circuits relay information to dictate worm behavior. The neurons in a circuit communicate with each other via chemical and electrical signals at specialized connections called synapses. Electrophysiological techniques are used to obtain a readout of synaptic events that elicit and modulate various behaviors. They have been used to complement genetic approaches in correlating behavior with specific genes and neurons. However, measuring electrical events at the worm synapse is challenging. The worm is about 1 mm long and the diameter of the neuronal cell bodies averages only 2-3 um, therefore, it is very difficult to gain access to the neurons. Also, the identification of cells or neurons of interest requires special measures [6]. The expression of fluorescent reporters in specific neurons and cell types has greatly aided the identification process [6]. The worms have a tough cuticle, which helps to maintain a high internal hydrostatic pressure, and piercing the cuticle disrupts the internal pressure severing neuronal connections [45]. Recent improvements in dissection methods that preserve the integrity of the worm have been an important advance in this area. Better techniques for permeabilizing the membranes and low-resistance patch pipettes have further facilitated the process of obtaining reproducible recordings.
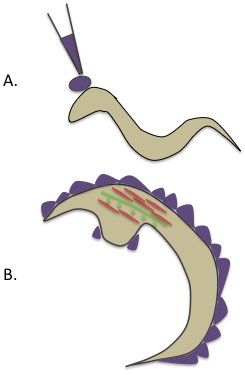
For recording electrical activity in worms, standard patch-clamp techniques can be applied. After the worm has been glued, a sharp glass needle is used to puncture the worm cuticle away from the recording site and to release hydrostatic pressure. The patch pipet is brought in contact with the membrane, and application of mild suction causes the cell membrane to enter the pipet. As the pipet resistance increases, a high resistance seal called the ‘gigohm seal’ is produced, which provides mechanical stability and helps in recording electrical activity with very little competing noise [6]. Adpated from [7].
Due to its well-defined anatomy and transparency, optical approaches have been successfully applied in worms to address fundamentally important questions in cell biology. In particular, studies relating to the development, organization and function of the nervous system have benefited immensely from these approaches. The worm nervous system is comprised of 302 neurons, which make about 7000 chemical and electrical synapses. C. elegans was the first model system in which GFP was used as a visual reporter [46], and in recent years, GFP-tagged reporters have been used extensively to study synaptic terminals and synaptic activity [47] or specific neurons such as eight dopaminergic neurons for Parkinson's disease research [48]. A presynaptic terminal, or synaptic bouton, is a specialized area in the axon that contains membrane-bound synaptic vesicles. The synaptic vesicles release neurotransmitters when an appropriate stimulus is received. The neurotransmitters bind to the receptors on the postsynaptic terminal, which transforms the signal (received via the neurotransmitter) into a chemical or electrical signal in the neuron or the muscle. The imaging of the worm nerve cord with GFP reporters has been used for quantitation of pre-synaptic proteins at the synapse. This has been possible because the amount of fluorescence, under ideal circumstances, is proportional to the concentration of the protein [49]. When synaptic architecture is disrupted or when neuron function is perturbed, the abundance and the distribution of synaptic proteins may change, which can be monitored by changes in the localization and fluorescence of the GFP-tagged reporter.
An important issue to consider in these experiments is that the spectral behavior of GFP is affected at low pH. GFP carries a single protonation site whose pKa is about 6. When GFP-tagged reporters are routed to acidic compartments, such as endosomes, lysosomes and synaptic vesicles, the GFP is protonated and the fluorescence is quenched [49]. Miesenbock and colleagues used a random mutagenesis strategy to generate a GFP variant called ‘pHluorin’ that has enhanced pH sensitivity and pKa of 7.1 [50]. This variant has been successfully used to monitor defects in neurotransmitter release and uptake at the synapse [51].
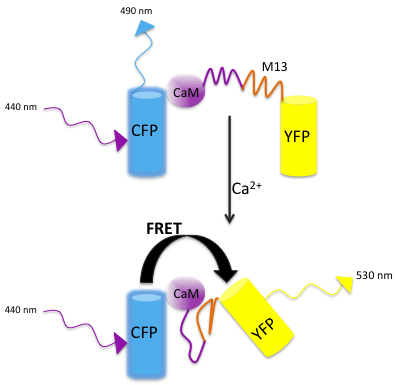
Calcium is a very important second messenger, therefore, considerable effort has been devoted to the development of tools that can efficiently and accurately monitor changes in intracellular calcium. The probes for monitoring calcium fall into two broad classes: calcium dyes and genetically encoded calcium indicators (GECI). Calcium dyes have been used extensively to monitor changes in neuronal activity that result from calcium signaling in live cells and in real time [52]. Many of these dyes work by chelating calcium that results in a change in their fluorescence spectrum. However, the calcium dyes have been used with very limited success in worms via direct injection [53]. The major limitation of using calcium dyes is the worm’s small size, which makes it difficult to introduce the dye directly into the tissue of interest. The use of GECIs allows researchers to circumvent this problem. The GECIs consist of a light-emitting component, a fluorescent protein, such as GFP or its variants, fused to a calcium-responsive element, such as calmodulin. The binding of calcium to calmodulin changes the spectral properties of the single chromophore and generates an optical readout. Another class of GECIs consists of calmodulin inserted between two fluorescent proteins, CFP and YFP. Calcium-induced changes alter the distance and the relative orientation of the fluorescent molecules, thereby altering the efficiency of fluorescence resonance energy transfer (FRET). For a ratiometric analysis, the fluorescence intensity of the donor CFP following excitation and the fluorescence intensity of the acceptor YFP upon donor excitation are captured [52]. The fluorescence ratio reflects the concentration of calcium and is directly proportional to it. It is important to optimize the parameters for data collection to obtain good signal to noise ratio and to minimize the photobleaching of sensors. The GECIs are non-invasive and these chimeras can be expressed in any organelle using signal sequences and tissue-specific promoters. Some disadvantages of using fluorescent probes should be kept in mind. The presence of the probe may alter the behavior of the worm. If the probe is expressed at high concentrations, it may buffer intracellular calcium and perturb calcium-dependent signaling. On the other hand, low expression of the probe may limit the acquisition of enough fluorescence signal. In addition, the ionic environment of the cell may influence the behavior of the probe. The continued efforts to improve the dynamic range, response kinetics and the effect of the cellular environment have expanded the repertoire of tools available for calcium imaging. These indicators will, no doubt, continue to provide useful resources for investigating the worm nervous system.
Gong et al designed an activity-based high-throughput genetic screen for mutants defective in cold sensation in C. elegans [54]. They monitored the cooling-triggered calcium response in the intestine of live worms cooled in a real-time PCR thermal cycler by recording changes in the fluorescence of GCaMP from the sample in real time (real time calcium imaging by fluorescence microscopy). Consequently, they identified GLR-3, a kainate-type glutamate receptor homolog, as a cold receptor. To characterize the cold-receptor they used GCaMP-based calcium imaging system to monitor the calcium response of mammalian cell lines expressing C. elegans GLR-3 or the mouse, zebrafish and human homologue Gluk2 [54]. Zullo JM et al, on the other hand, measured neural excitation through GCaMP calcium imaging in the glutamatergic ASH neurons of worms [17].
Optical methods, calcium imaging and electrophysiological approaches have provided important insights into worm behavior. However, to truly understand and appreciate the neural circuits that dictate behavior, recordings must be made in freely behaving and moving worms. Using optical probes expressed in specific neurons, recordings have been made in untethered worms, however, this requires that the target be periodically recentered to keep the neurons of interest in view either manually or by image-processing software [55]. There are two major problems with this system: as the target moves rapidly, substantial blurring occurs leading to high signal to noise ratio, and that in freely moving worms, the target usually escapes the field of view while the image is being captured and processed and the microscope stage is being recentered. To overcome these limitations, Shawn Lockery’s group devised an image-free, optomechanical system that continuously recenters a fluorescent target moving at speeds of up to 500 m/sec while neuronal activity and behavior are being recorded [56]. To allow high resolution of neurons of interest in rapidly moving worms, this system is compatible with high-performance microscope objectives. When compared to previously used systems, the latency of recentering movements is dramatically reduced because this system uses a high-speed feedback loop between the location of the target in the field of view and the compensating movements of the stage. This system uses two light paths with low and high magnification. The path with lower magnification (usually a 4X objective) is used for monitoring the behavior and recording the image of the worm. The high magnification path (usually a 63X-100X objective) is used for calcium imaging and the actual tracking procedure. The system uses beam splitters to divert 20% of the light from a 63X objective to a four-quadrant photomultiplier tube (PMT) for calcium imaging. Therefore, the four analog signal intensities are used to regulate the speed of the motors on the motorized stage. As the light falling on the PMT is not converted into an image, the problem of it going out of focus and being lost is eliminated. The system can also be used for creating so-called ‘virtual environments’ by optogenetic activation of sensory neurons [56]. As the image-processing step for recentering the stage is completely removed, thus far, this is the fastest worm tracking system [57].
Studies with C. elegans have greatly advanced our knowledge of the way multicellular organisms function. The genetic screens have been critical in understanding and appreciating how biological pathways orchestrate development, learning, behavior and ageing. Future developments in transgene expression systems, whole genome analysis, RNAi interference and optical methods will continue to enrich our knowledge and understanding of C. elegans.
The effects of various chemicals on the development, survival and motion can be effectively evaluated using C. elegans. Both microwell plates and microfluidic devices are used for C. elegans-based high-throughput screening (HTS) of various chemical compounds. However, microfluidic analysis has been proven to be more precise for testing different drug concentrations. Several methods, such as laser axotomy, changeable C. elegans immobilization and imaging with high resolution, have been applied for HTS.
In particular, the targeting of the neuronal cells is performed by laser axotomy, supported by highly selective laser irradiation in order to prevent the destruction of nearby cells [58]. With regard to the immobilization techniques, on-chip methods have recently been developed in order to prevent suppression of C. elegans development during the analysis, when the worms are usually immobilized in microchambers [59]. These approaches include the spatial restriction of C. elegans [60] and the application of thermolabile gels [61].
Multiple studies using HTS based on microfluidics, which includes the analyses compound toxicity, behavior and calcium activity, have recently been published. For instance, a microfluidic device has been used to study the life span and motion pattern under oxidative stress [62]. This method was applied for testing the effects of anti-aging drugs. Toxicity screening of various chemicals has also been performed in C. elegans using microfluidics [63]. Remarkably, C. elegans shows high variability of behavioral patterns with regard to movement, light sensitivity and learning ability. Also, calcium activity in C. elegans neurons has been assessed by microfluidic compound activation [64, 65]. The motion patterns and sleep stage of the experimental worms were studied in microfluidic compartments and demonstrated the dopamine-mediated regulation of spatial distribution selectivity [66, 67].
The author would like to thank Yingsong Hao, Ph.D. and Kavita Babu, Ph.D. for comments and suggestions. Dr. Konstantin Yakimchuk added the section "Application of C. elegans for high-throughput drug screening" in May 2018.
- Gartner et al. Germline Survival and Apoptosis. 2008;WormBook, ed. The C. elegans Research Community, WormBook, , http://www.wormbook.org. Available from: dx.doi.org//10.1895/wormbook.1.145.1
- Strange K, editor. C. elegans methods and applications. Humana Press Inc., Totowa, NJ, USA; 2006.
- Richmond, J. E. Electrophysiological recordings from the neuromuscular junction of C. elegans. 2006;WormBook, ed. The C. elegans Research Community, WormBook, , http://www.wormbook.org. Available from: dx.doi.org//10.1895/wormbook.1.112.1
- Jorgensen E, Mango S. The art and design of genetic screens: caenorhabditis elegans. Nat Rev Genet. 2002;3:356-69 pubmed
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71-94 pubmed
- Hobert O. Behavioral plasticity in C. elegans: paradigms, circuits, genes. J Neurobiol. 2003;54:203-23 pubmed
- Riddle DL, Blumenthal T, Meyer BJ, and Priess JR, editors. C. elegans: A Practical Approach. 2nd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997.
- Grishok A, Mello C. RNAi (Nematodes: Caenorhabditis elegans). Adv Genet. 2002;46:339-60 pubmed
- Kamath R, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313-21 pubmed
- Kennedy S, Wang D, Ruvkun G. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature. 2004;427:645-9 pubmed
- Evans TC, editor. Transformation and microinjection. Wormbook, ed. The C. elegans Research Community. , http://www.wormbook.org; 2006. Available from: dx.doi.org//10.1895/wormbook.1.108.1
- Jin, Y. Transformation. In: Hope, IA, editor. C elegans: A Practical Approach. Oxford University Press; New York; 1999.
- Mello C, Kramer J, Stinchcomb D, Ambros V. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959-70 pubmed
- Kelly W, Xu S, Montgomery M, Fire A. Distinct requirements for somatic and germline expression of a generally expressed Caernorhabditis elegans gene. Genetics. 1997;146:227-38 pubmed
- Merritt C, Gallo CM, Rasoloson D, Seydoux G. Transgenic solutions for the germline. Wormbook, ed. The C. elegans Research Community. , http://www.wormbook.org; 2010. Available from: dx.doi.org//10.1895/wormbook.1.148.1
- Praitis V, Casey E, Collar D, Austin J. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics. 2001;157:1217-26 pubmed
- Giles N, Geever R, Asch D, Avalos J, Case M. The Wilhelmine E. Key 1989 invitational lecture. Organization and regulation of the qa (quinic acid) genes in Neurospora crassa and other fungi. J Hered. 1991;82:1-7 pubmed
- Huiet L, Giles N. The qa repressor gene of Neurospora crassa: wild-type and mutant nucleotide sequences. Proc Natl Acad Sci U S A. 1986;83:3381-5 pubmed
- Francis M, Mellem J, Maricq A. Bridging the gap between genes and behavior: recent advances in the electrophysiological analysis of neural function in Caenorhabditis elegans. Trends Neurosci. 2003;26:90-9 pubmed
- Chalfie M, Tu Y, Euskirchen G, Ward W, Prasher D. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802-5 pubmed
- Nonet M. Visualization of synaptic specializations in live C. elegans with synaptic vesicle protein-GFP fusions. J Neurosci Methods. 1999;89:33-40 pubmed
- Miesenbock G, De Angelis D, Rothman J. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192-5 pubmed
- Dittman J, Kaplan J. Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proc Natl Acad Sci U S A. 2006;103:11399-404 pubmed
- Dal Santo P, Logan M, Chisholm A, Jorgensen E. The inositol trisphosphate receptor regulates a 50-second behavioral rhythm in C. elegans. Cell. 1999;98:757-67 pubmed
- Clark D, Gabel C, Gabel H, Samuel A. Temporal activity patterns in thermosensory neurons of freely moving Caenorhabditis elegans encode spatial thermal gradients. J Neurosci. 2007;27:6083-90 pubmed
- Husson SJ, Wagner SC, Schmitt C, Gottschalk. Keeping track of worm trackers. Wormbook, ed. The C. elegans Research Community., http://www.wormbook.org; 2012. Available from: dx.doi.org//10.1895/wormbook.1.150.1
- Materials and Methods [ISSN : 2329-5139] is a unique online journal with regularly updated review articles on laboratory materials and methods. If you are interested in contributing a manuscript or suggesting a topic, please leave us feedback.
- method
- Adenoviral Vectors
- Behavioral Phenotyping in Rats and Mice
- CRISPR and Genomic Engineering
- GFP Antibody
- Laboratory Mice and Rats
- Neuronal Activity Research Methods
- Nucleic Acid Delivery: Lentiviral and Retroviral Vectors
- Stem Cell Research Using Mouse Models
- Zebrafish Research Methods
- siRNAs and shRNAs: Tools for Protein Knockdown by Gene Silencing