This article reviews the main methodologies for genomic engineering, focusing on the CRISPR/Cas 9 system.
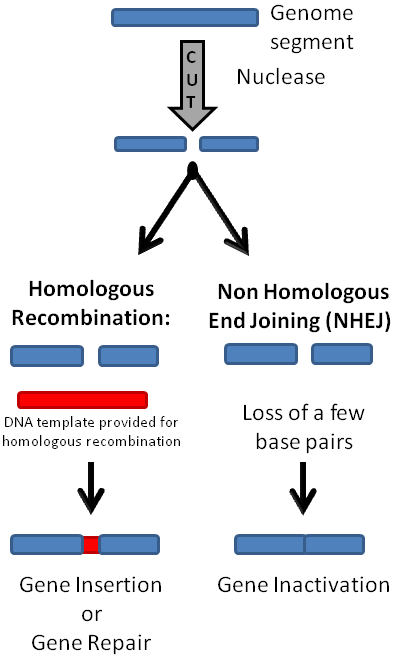
This article reviews the main methodologies for genomic engineering, focusing on the CRISPR/Cas 9 system. Genome editing technologies aim to make small changes in an organism’s own genes in order to modify a specific characteristic. And while gene editing occurs naturally, cutting edge methodologies of these processes allow for immediate applications to human health and also to plant and animal agriculture. Genome editing employ proteins/enzymes and or RNA sequences that bind to specific DNA sequences, and target adjoined nucleases to the genome. In general, the nucleases introduce double-strand DNA breaks (DSB) in the target nucleic acid sequence. After a DSB occurs, the cell’s own DNA repair mechanisms mend the breaks. Repair mechanisms include homologous recombination, which requires a homologous template for repair but the template may be endogenously or exogenously supplied (Figure 1). The DSB may also be repaired by a mechanism that does not need a template, such as nonhomologous end joining (NHEJ) (Figure 1) or microhomology-mediated end joining (MMEJ) [3]. These cellular DNA repair mechanisms are utilized for different gene editing goals. NHEJ is more useful for excising part of the genome or inactivating a gene, while homologous recombination with an exogenously supplied template can be utilized to create gene insertions or make base pair substitutions, and is more applicable for repairing genetic mutations. We also review the development of transfer DNA (T-DNA) binary vector system which facilitated the advancement of plant-based genomic editing for agricultural purposes.
There are several successful approaches for targeted genome editing [4]. Meganucleases are homing endonucleases discovered in yeast that recognize fairly long DNA sequences, and create double-strand breaks that are mended via stimulation of homologous recombination [5]. Other genome editing systems utilize the endogenous DNA repair mechanism of nonhomologous end joining. These include nucleases coupled to either zinc finger binding protein domains, which are used to create zinc finger nucleases (ZFN) [6], or transcription activator-like effector (TALE) domains, which are linked to nucleases to create TALENs [7]. Although these systems are effective and commercially available, they are somewhat costly and time-consuming, since they require the design of new DNA binding protein domains for each new DNA target. The newest genome editing approach employs the prokaryotic immune defense system based on clustered regularly interspaced short palindromic repeats (CRISPR, pronounced crisper), and CRISPR-associated genes (Cas) [8, 9]. This system relies on complementary base pairing between RNA from the CRISPR loci and the target nucleic acid sequence [10]. A considerable advantage of the CRISPR system over the previous DNA binding protein methodologies is that it utilizes an RNA molecule to target the nuclease portion of the system to a specific nucleic acid target, and RNA is much easier and cheaper to synthesize than DNA binding proteins. Although genomic engineering has great potential in curing genetic diseases, a group of prominent scientists urge a cautious approach in its application in human germlines [11], especially because any off-target DNA damage may give rise to significant cellular toxicities. Genome editing techniques, especially CRISPR, have been adopted for diagnostics purposes [12].
CRISPR are genomic loci composed of short DNA repeats with spacer sections interspersed (Figure 2). Originally observed in E. coli [8], these loci have been found in a variety of Archaea and bacterial genomes. CRISPR loci acquire fragments of foreign DNA from phages or plasmids, which are inserted within their repeat sequences [13]. The foreign integrated DNA sequences aid the cell in recognizing and targeting foreign DNA in future attacks [14]. That CRISPR-Cas systems work via RNA interference was first proposed by Makarova et al in 2006 [13], and later supported by many reports, and reviewed by members of the same group [15].

CRISPR loci include repeats, spacers, a leader sequence and CRISPR-associatedg enes (Cas) [9]. There are several different Cas genes, which produce proteins with varying functions including endonuclease, helicase, nucleic acid binding and transcription regulation [16]. CRISPR-Cas systems are classified into several types [17]. Type II CRISPR systems are the simplest and have the fewest number of associated genes, which include the nuclease cas9, as well as the proteins cas1, and cas2. Some Cas proteins process the CRISPR gene product [9] into mature CRISPR RNA (crRNA) and transactivating crRNA (tracrRNA) molecules. The crRNA is complementary to a target sequence, normally a foreign nucleic acid segment derived from a virus or conjugative plasmid. Together, CRISPR RNAs and Cas proteins form a complex that recognizes the foreign or target nucleic acid sequence and causes its degradation. This system provides RNA-based immunity against foreign DNA [18-20]. CRISPR-Cas systems can lead to the degradation of both target DNA and target RNA, depending on the CRISPR loci and the type of Cas proteins involved [21, 22]. More recently, P Pausch et al identified a single 70 kD CasΦ protein using the same active site for both CRISPR RNA processing and crRNA-guided DNA cutting from bacteriophages [23].
The Type II CRISPR/Cas system has been utilized for directing sequence-specific targeting of novel nucleic acid targets. This system includes the Cas9 nuclease, a non-coding tracrRNA and a pre-crRNA array that includes sequences that guide nuclease targeting. It was reported by Jinek et al that the Cas9 nuclease can be guided to a novel DNA target for cleavage by engineering a synthetic guide RNA (sgRNA), which replaces the two RNAs normally found in the CRISPR-Cas system (CRISPR RNA or crRNA and transactivating crRNA or tracrRNA) [10]. In January 2013, publications in Science by the Zhang [24] and Church [25] research groups reported their successful use of an RNA-guided endonuclease in mammalian cells. Cong et al reported Cas9 mediated cleavage of endogenous genomic sites in both mouse and human cells [24]. In addition, they reported that Cas9 could also function as a nickase, an enzyme capable of introducing a single-strand cut with the same specificity as a regular CRISPR/Cas9 nuclease [26]. Mali et al used a custom guide RNA to target the Cas9 enzyme to the AAVS1 locus in a variety of human cells, and reported a computed resource of nearly two hundred thousand unique guide RNA sequences that target about 40% of the human genome’s exons [25]. Both groups utilized multiple synthetic RNAs at once to target the cleavage of several loci simultaneously.
Jinek et al have reported their success with a modified Cas9 protein for promoting NHEJ in human cells [27]. Hwang et al have demonstrated their success with the CRISPR-based sgRNA-Cas9 approach to carry out genome editing in zebrafish embryos, which induced modifications at a similar rate to that found when using ZFNs and TALENs [28]. Interestingly, in bacteria at least, the CRISPR-Cas9 system will direct nuclease induced DSBs even with a minor mismatch in the 5’ end of the target sequence [29] and also in mammalian cells [24, 25]. Cho et al targeted the HIV receptor gene CCR5 in a human cell line utilizing a CRISPR-Cas9 system [30]. The application of a CRISPR-Cas9 based system to excise the proviral HIV-1 DNA in the human genome and to prevent new HIV-1 infection has been reported [31].
A considerable advantage of the CRISPR-Cas9 system over the previous DNA binding protein methodologies is that it utilizes an RNA molecule to target the nuclease of the system to a specific nucleic acid sequence. RNA is easier and cheaper to synthesize than the protein domains used with the ZFN and TALEN approaches. In addition, the gRNA sequences can be validated through mismatch-cleavage SURVEYOR assay [32]. Applications of CRISPR-Cas9 systems are as varied as those for ZFNs, TALENs and meganucleases. For example, CRISPR systems can be utilized to engineer phage resistant bacteria. Recently, a group has reported their success with utilizing a CRISPR-Cas system to engineer site-specific mutagenesis and allelic replacement in yeast [33]. More applications and technologies based on the systems described above are already being reported [34]. Qi et al described an inactive Cas9 protein, that, when expressed with a synthetic guide RNA, creates a recognition complex that interferes with transcriptional elongation and successful binding to DNA targets by RNA Polymerase or transcription factors [35]. This system is called CRISPR interference (CRISPRi), and it can be utilized to repress the expression of multiple genes at once in a variety of experimental systems [36].
The large number of recent publications on CRISPR–Cas9 reflects the effectiveness and popularity of this genome-editing tool. Cell-selective gene editing has been achieved through the engineering of S. pyogenes Cas9 proteins harboring asialoglycoprotein receptor ligands (Cas9-ASGPrL) and delivery of ribonucleoproteins by its cognate receptor [37]. Genomic modifications by CRISPR–Cas9 have been reported to cause p53-mediated DNA damage and cell cycle arrest [38]. The authors have pointed out that p53 inhibition might elevate the efficiency of genome editing and emphasized the importance of p53 monitoring for the generation of cell-based therapeutic approaches. In addition, CRISPR–Cas9 has been applied to investigate the functions of various regulatory proteins, such as β-arrestins [39]. The study found that β-arrestins control G-protein coupled receptors and ERK1/2 signaling pathways.
With regard to on-target mutagenesis, CRISPR–Cas9 has been used to induce large deletions at the target regions in both mouse and human cells [40]. The study also found that DNA breaks generated by RNA/Cas9 often caused extended deletions and crossover recombination, which might have pathological outcomes. Wienert B et al established a procedure, DISCOVER-Seq, to detect CRISPR off-targets in vivo [41].
CRISPR-Cas9 system resources include several websites and Google Groups as outlined in Table 1. Dr. Zhang, one of the CRISPER-Cas9 leaders established a website full of valuable information about utilizing CRISPR methods in research ( zlab.bio/resources). ChopChop is a common sgRNA design tool [42, 43]. Liu Y et al designed sgRNA targeting sequences using the MIT online tool [44]. The site also predicts potential off-target sites, which can be used for further examinations, for example, [45]. Vaidyanathan S et al used a different website (COSMID Off-Target Analysis Tool) at https://crispr.bme.gatech.edu/ [46]. CasOFFinder can also be used to predict the off-target sites, which can be further examined [47, 48]. In addition to CasOFFinder, SITE-Seq is commonly used to examine the off-target editing [48, 49].
Both commercial and non-profit organizations provides ready-made Cas9 vectors, such as Alt-R CRISPR-Cas9 from Integrated DNA Technologies [50], GeneArt CRISPR Nuclease Vector Kit from Life Technologies [51], LentiCRISPR vector V2 52961 [50, 52], human codon-optimized SpCas9 and chimeric guide RNA expression plasmid pX330-U6-Chimeric_BB-CBh-hSpCas9 ( 42230) [53], pGL3-U6-sgRNA expression vector [44], LentiCas9-Blast vector ( 52962) [54, 55], pSpCas9(BB)-2A-GFP (PX458) ( 48138 [56-58] ), and pSpCas9(BB)−2A-Puro (PX459) ( 48139 [59] ; version 2 62988 [60, 61] ) from Addgene, CMV::Cas9-2A-eGFP vector (CAS9GFPP-1EA) [62] and lentivirus-based CRISPR/Cas9 plasmids pLV-U6g-EPCG-Cav1 and pLV-U6g-EPCG-Cltc [63] from Sigma. Cas9-knockin mice with global expression are also available from academic source [64], which enables specific mutations with simple introduction of sgRNA. Yang J et al cloned guide RNAs into the PX458-mCherry vector to disrupt PAC genes in HEK293 and HeLa cells and generate PAC knockout mice [56]. PX458 vectors enable easy sorting of transfected GFP+ cells. He M et al used pLenti-Cas9-Blast and pLKO-gRNA-Puro from GenScript and Xfect mESC transfection reagent from Clontech to knockout multiple genes such as Rnf2 in ES cells [65]. Laflamme C et al introduced Cas9 into U2OS first with pAAVS1-PDi-CRISPRn knockin vector and AAVS1 TALEN pair, and then transfected sgRNAs against C9ORF72 into the stable-inducible Cas9 U2OS cells using JetPRIME transfection reagent to generate KO U2OS cellline [61]. Huang H et al developed inducible METTL14-knockout HepG2 cells using lentiviral vectors for Cas9 (pTOL-hCMVTET3G-Hygro and pCLIP-Cas9-Nuclease-TRE3G-ZsGr) and sgRNA (pCLIP-hCMVgRNA-RFP) expression from transOMIC Technologies [57].
Ready-made KO plasmid kits with gRNAs are available commercially as well. Rigau M et al generated knockout cell lines with the BTN3A1 CRISPR-Cas9 KO Plasmid kit from Santa Cruz Biotechnology [51].
Cell lines with inducible Cas9 expression can provide a great platform to introduce new KO quickly and the efficiency of indel generation can be assessed through a reporter vector. For example, Feldman D et al selected the clones from parental HeLa-TetR-Cas9 cells with the reporter vector expressing GFP and an sgRNA targeting GFP pXPR_011 (Addgene #59702) [66].
JetPEI reagents from Polyplus has also been used to transfect RAW264.7 cells with sgRNAs and Cas9 expression plasmids [44]. Its in vivo-jetPEI was used to directly introduce sgRNA expression vector and S. pyogenes dCas9 expression vector into mouse brains to knock out lncRNA Neat1 [67].
Direct introduction, through electroporation [68] or lipofectamine treatment or injection, of sgRNA and Cas9 nuclease can provide a quick approach to knockout certain genes. B Shen et al injected CleanCap Cas9 mRNA from TriLink and sgRNAs (transcribed using Ambion MEGAshortscript Kit and purified using the Ambion MEGAclear Kit) into C57BL/Ka zygotes to generate Oln mice [69]. IA Klein et al transfected V6.5 murine embryonic stem cells and HCT116 cells with a Cas9 plasmid and non-linearized homology repair templates using Lipofectamine 3000 from Invitrogen (L3000) [70]. Dai H et al injected sgRNA and Cas9 mRNAs into C57BL/6J pronuclear-stage zygotes to remove the Pira gene cluster and to remove a segment of Pirb gene (both of them are duplicated on chromosomes) to generate Pira- and Pirb-deficient mice [71]. Cunrath O and Bumann D injected in-vitro transcribed sgRNA, ssDNA homologous recombination template and Cas9 protein from Toolgen into fertilized mouse oocytes to generate SLC11A1-deficient mice [45]. Zhang J et al injected Cas9 mRNA and gene-specific guide-RNA (sgRNA) into mouse zygotes to generate a number of knockout and knockin mice to investigate sour sensing [72]. Samir P et al injected sgRNAs, Cas9 mRNA transcripts, and homology-directed repair oligonucleotides into pronuclear-stage mouse zygotes to generate transgenic mice [73]. Boettcher S et al electroporated recombinant Cas9 protein, synthetic crRNA and tracrRNA into K562 and MOLM13 cells to generate TP53 mutations or knockouts using a 4D-Nucleofector from Lonza [74]. Vodnala SK et al electroporated synthetic sgRNA (control or against ATG7) and Cas9 nuclease into activated T cells with Thermo Fisher Neon transfection [75]. OPTI-MEM buffer is reported to be the best media for electroporation [46]. Chopra S et al introduced sgRNA-CAS9 ribonucleoprotein complexes with lipofectamine CRISPRMAX transfection reagent from Invitrogen into human monocyte–derived dendritic cells to knockout XBP1 and ERN1 genes [76].
Guo CJ et al devised a novel CRISPR-Cas9–based genetic system for Clostridium sporogenes using conjugation and a two-vector design to overcome the inefficiency of homologous recombination in Clostridium species [77].
CRISPR-Cas9 knockout screens can quickly identify genes that are involved in specific phenotyptes, such as those genes that inhibited or promoted copper-induced cell death [78], SUMO1 degradation [79], DNA methylation [80] or the formation of lipid droplets in BV2 cells [81].
Transcription activator-like effectors (TALEs) are DNA-binding domains that can be linked together modularly and fused with nuclease domains to create TALE nucleases, or TALENs [82]. Each TALE has a repeat of 34 amino acids and recognizes a single base pair on the DNA target molecule. Therefore, several TALEs must be linked together to create a protein that recognizes a specific DNA sequence. As with ZFNs, two arrays are used to target a nuclease to a DNA sequence, and each array binds one half-site target (Figure 3). TALE arrays are primarily fused to Fok1 nuclease domains, which cleave DNA within the spacer region between the two half-sites. ZFN and TALENS are both modular and have natural DNA-binding specificities. They differ in that ZFN's single zinc finger domains recognize three base pairs, and TALE domains recognize just one base pair. This difference does make it easier to create TALEN systems that recognize more target sequences. One benefit of TALENs over ZFN’s for genome editing is that they exhibit less toxicity in human cell lines and zebrafish [83]. Another benefit of TALENs is a higher rate of genome editing success via cytoplasmic injection of TALEN mRNA in livestock embryos than occurs with ZFN induction [84].
Applications of TALENs include the creation of genetically modified mice, which may be used as a model for human disease research. For instance Sung et al utilized a TALEN based approach to create a mouse with a genetic knockout of the progesterone immunomodulatory binding factor 1 gene [85]. In human stem cells, TALENS have been used to create various disease states including insulin resistance and hypoglycemia, which can be used as a model to study the disease [86]. TALENS have been well utilized in plant genome engineering [87], and in conditional genome editing in C. elegans [88].
Although current genomic engineering favors CRISPR methods, TALENs may still be of a primary choice in some experimental species. For instance, TALENs have been used for targeted genome editing in Xenopus tropicalis by knocking out Klf4 [89] or thyroid hormone receptor α [90]. In mice, TALEN-induced indel generation in the hemoglobin promoter induced high expression of hemoglobin in vivo [91].
In addition, TALENs have been applied for genomic modifications in human stem cells. Lee J et al introduced multiple changes to the LMNA gene in iPSC lines through TALEN-mediated genome editing to study the role of LMNA mutations in dilated cardiomyopathy [92]. A Talen-mediated approach has recently been used to induce a p53 mutation in the human embryonic stem cells. The obtained cell line was characterized by pluripotency and the ability to generate a teratoma, which contained tissues of all three germ layers [93]. Furthermore, TALEN technique has been applied to generate amniotic mesenchymal stem cells overexpressing anti-fibrotic IL-10 [94].
Several resources for purchasing or making your own TALENs are available (Table 1). Yasuda S et al generated knockin HCT116 cells with Platinum Gate TALEN Kit (Addgene 1000000043) and TALE nuclease (TALEN) Kit (Addgene 1000000019) [60]. TALENs are commercially available from Cellectis Bioresearch and Life Technologies. Modular assembly options include the Voytas Kit [82], and the Zhang kit [95]. These kits utilize various approaches to create TALE libraries and arrays. FLASH Assembly is a fast, high-throughput assembly system of creating custom TALENs using a library of premade TALE arrays [96]. A do-it-yourself protocol for assembling TALENS within mammalian expression vectors in just 2 days was published recently [97]. Another group reported their creation of a large library of TALEN plasmids for over 18,000 human protein-coding genes [98].
Meganucleases (MGN) are sequence-specific endonucleases with large cleavage sites (14–25 bp) that can create double-stranded breaks at specific locations. Meganucleases are encoded by mobile genetic elements that induce recombination in a process called homing. Some meganucleases tolerate small homing site sequence discrepancies, but the large recognitions sites still provide these enzymes with a high degree of specificity to a particular DNA target sequence, which in turn establishes a low level of off-target cleavage within a genome and low toxicity. Homing endonucleases are naturally occurring mobile DNA sequences within several organisms, including bacteria, fungi and some plants. There are hundreds identified to date. Meganucleases are coded for by open reading frames within a mobile sequence inside a self-splicing RNA intron or self-splicing protein, intein. The sequence that the meganuclease recognizes is referred to as its homing site, and is made of DNA pieces from the two intron or intein flanks [99]. Homing endonucleases recognize and cleave the intronless cognate allele, initiating the homing event, which is a transfer of an intervening sequence to the homologous allele that lacks the sequence. When a DNA sequence is cleaved by a homing endonuclease, it can be repaired by homologous recombination. This results in the transfer of the homing endonuclease-containing intron and disruption of the homing site sequence.
There are several families of homing endonucleases and their specificity and catalytic mechanisms are well reviewed by Chevalier and Stoddard [99]. The LAGLIDADG motif family of homing endonucleases is the most frequently used in genetic engineering. Commonly utilized LAGLIDADG family members include I-SceI and I-CreI. This family of homing endonucleases includes proteins with 1 or 2 copies of the LAGLIDADG motif. Enzymes with only one copy of the motif function as a homodimer that recognizes almost palindromic homing sites, and include family members I-CreI and I-CeuI. Enzymes with two copies of the LAGLIDADG motif separated by about 100 residues act as monomers and include family members such as I-DMO1 and Pi-SceI.
The very large recognition sites of meganucleases provide them with great specificity, which decreases the potential of toxicity to cells and organisms. A major limitation of the use of naturally occurring meganucleases is that the large recognition sites may exist only once in an organism's genome just once, or possibly not at all. If no naturally occurring DNA homing site is prosent within the genome, one must introduce a target sequence prior to utilizing a homing endonuclease to cleave the DNA. In order to utilize meganucleases at selected DNA sequences, scientists have turned to modifying the recognition sites of existing meganucleases by mutagenesis or by the creation of chimeric enzymes. For example, Seligman et al described mutating the I-CreI meganuclease recognition site [100].
Meganucleases have several potential applications. Due to their high degree of specificity, they offer an advantage over less specific genetic editing tools for the development of disease therapies. Homing endonucleases have the potential to be developed into therapies for a multitude of inherited diseases caused by frameshift mutations or nonsense codons [101]. M Aubert et al found that meganucleases delivered through adeno-associated virus, but not CRISPR/Cas9, could efficiently eliminate latent Herpes simplex virus from superior cervical ganglia [102]. Treatments for Duchene muscular dystrophy have also been based on meganucleases, which can restore the reading frame of a mutated dystrophin gene [103]. Utilization of meganucleases for gene therapy applications has been well reviewed recently by Silva et al [104].
Scientists can utilize meganucleases for genome editing by either purchasing naturally occurring or pre-engineered meganucleases from a commercial supplier such as Cellectis. There are also databases dedicated to engineering LAGLIDADG homing endonucleases [105]. Refer to Table 1 for links to these resources.

Zinc finger nucleases (ZFN) are manufactured proteins with zinc finger domains that recognize a DNA sequence fused to a nuclease domain (often from the bacterial restriction enzyme FokI). The zinc finger domains are DNA binding domains that each recognizes a 3 bp DNA sequence. Several zinc finger domains are joined to create an array of DNA binding domains that will recognize a particular DNA target, and two arrays are used to target 2 nuclease domains to the sequence between the two arrays. Array length affects ZFN activity. One study found that six-finger arrays led to successful DSBs in 71% of the targeted loci as opposed to previous success rates of only 25% with shorter array lengths [106]. ZFNs create double-strand breaks (DSBs) in the DNA sequence, which is repaired by the endogenous repair machinery of the cell via either nonhomologous end joining (NHEJ) (Figure 3) or homologous recombination. The zinc finger portion of a ZFN can be engineered to recognize many DNA sequences of interest specifically. Unfortunately, ZFNs do produce some overt toxicity to cells due to off-target cleavages.
An exciting application of ZFNs is the mutation of zebrafish genes. Foley et al recently described their use of the Oligomerized Pool ENgineering method (OPEN) to generate ZFNs that work well in zebrafish [107]. Another group has utilized ZFNs to repair the nucleotide sequence of the mutated Duchene gene and have demonstrated in mammalian cells that the ZFN treatment induces expression of the normal protein [108]. Yet another application of ZFNs is the generation of HIV-resistant T cells ex-vivo [109]. Duncan A et al deleted a 169-base-pair region from Tcf7l2 gene in rats through zinc-finger nucleases [110].
A zinc-finger domain array can also be linked to a transcription repressor domain to suppress the expression of a gene, in circumstances that CRISPR lacks the selectivity. Zeitler B et al, for example, tested such a construct to depress the expression of the huntington gene with CAG expansion in a mouse Huntington's model and human neurons [111].
Resources for obtaining ZFNs for particular DNA targets include both commercial vendors and several methodologies as recently described in detail by Perez-Pinera et al [112] (Table 1). A major supplier of ZFNs for custom targets is Sigma-Aldrich’s CompoZr Zinc Finger Nuclease service, which is partnered with Sangamo Biosciences and utilizes its proprietary method for creating ZNFs. Another resource is called Zinc Finger Tools, which is an open-access website that allows users to search for target sites from a DNA sequence of interest for nuclease targeting [6]. This site offers scientists a database of characterized zinc finger domains and a reverse engineering option that predicts binding sites for known zinc finger proteins. The Zinc Finger Consortium is yet another resource for those looking to start working with genome editing using zinc finger nucleases. This site includes publicly available methods for engineering zinc finger domains including Context-dependent Assembly (CoDA) [113], Oligomerized Pool Engineering (OPEN) [114], and Modular Assembly [106].

In order to improve targeting of specific sequences with ZFNs, a set of architectures, which provide the configurational options, has recently been developed [115]. The authors have demonstrated that the generated architectures may be effectively applied to modify various loci for therapeutic purposes. Also, antitumor effects were significantly activated by a combination of Zinc Finger Nuclease-mediated downregulation of HPV16/18E7 with Cisplatin and Trichostatin in cervical tumor cells [116].
More applications and technologies based on the systems described above are already being reported. The type II CRISPR/Cas9 system was adapted for transient gene expression regulation. By fusing of a transcription repression (KRAB) or activation (VP64) module to a mutant, inactive, Cas9 the CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) system (Figure 4) were generated [117]. The two systems already proved to be useful research tools with great perspectives in clinical applications. Multiple comprehensive reviews on both aspects were published recently. Here we exemplify just a few: [118-121]. Interesting comparative analyses of CRISPRi and the widely used RNA interference system are also available. Two of them can be found here [122, 123].
| Meganucleases | Zinc Finger Nucleases | TALENs | CRISPR/Cas9 | |
|---|---|---|---|---|
| DNA-recognition mechanism | Protein-DNA interactions through HR | Protein-DNA interactions that introduce DSB | Protein-DNA interactions that introduce DSB | RNA-guided protein-DNA interactions that introduce DSB |
| Length of DNA-recognition site | >14 | 18-24 | 20-59 | 20-22 |
| Target specificity | High Only some positional mismatches are tolerated Re-targeting requires protein engineering | High G-rich sequence preference Only some positional mismatches are tolerated Re-targeting requires protein engineering | High Requires a T at each 5’-end of its target Some positional mismatches are tolerated Re-targeting requires complex molecular cloning | Moderate RNA-targeted sequence must precede the 2 base pairs recognized by PAM Only some positional mismatches are tolerated Re-targeting requires new RNA guide Protein engineering is not required |
| Multiplexing | Not easy | Not easy | Not easy | Easy |
| Delivery | Easy via electroporation and viral vectors transduction | Easy via electroporation and viral vectors transduction | Easy in vitro delivery difficult in vivo due to the large size of TALEN DNA and their high probability of recombination | Easy in vitro Moderate difficulty of delivery in vivo due to poor packaging of the large Cas9 by viral vectors. |
| Use as gene activator | No | Yes Activation of endogenous genes Minimal off-target effects May require engineering to target particular sequences | Yes Activation of endogenous genes Minimal off-target effects No sequence limitations | Yes Activation of endogenous genes Minimal off-target effects Requires “NGG” PAM next to the target sequence |
| Use as gene inhibitor | No | Yes Works by blocking transcription elongation via chromatin repression Minimal off-target effects May require engineering to target particular sequences | Yes Works by blocking transcription elongation via chromatin repression Minimal off-target effects No sequence limitations | Yes Works by blocking transcription elongation via chromatin repression Minimal off-target effects Requires “NGG” PAM next to target sequence |
| Cost | High | High | High | Reasonable |
| Popularity | Low | Low | Moderate | High |
| Online resources | Database and Engineering Server for LAGLIDADG Homing Endonucleases | The Zinc Finger Consortium includes software tools and protocols ZFNGenome – resources for locating ZFN target sites | Mojo Hand [89] or E-TALEN [124] for TALEN design CHOPCHOP target site selection [125] | Guide design: Zlab, CRISPOR), Benchling [58] Other CRISPR resources: AddGene |
In addition, secondary techniques aimed to optimize the use of engineered nucleases in genome editing are also under development. For instance, recently enrichment strategies based on antibiotics and magnetic separation [127] or on the use of surrogate reporters [128] have recently been reported. Also, researchers continue to search for and to develop novel nucleases. Novel monomeric nucleases composed of a site-specific meganuclease cleavage head with additional affinity and specificity provided by a TAL effector DNA binding domain, the MegaTALs were developed. Boissel and Sharenberg described the process of assembling a MegaTAL, as well as a method for the characterization of its nuclease activity in vivo [129]. A multi-reporter system for the design and selection of highly specific novel nucleases was recently described by Oakes et al [130].

One of the main goals in utilizing genome editing in plants is crop improvement to advance the yield-related trait; however, several limitations in regard to the specificity in the genetic modificatios and the incompatibility of the host species have limited the development of crop improvements [4]. Gene editing using site-directed nucleases is one of the important potential techniques that helped overcome some of these limitations by specifically targeting a compatible region in a gene/genome. Applications of the gene editing techniques, including those discussed above, appear to be endless ever since their discovery and several modifications in original technologies have further brought precision and accuracy in these methods. Together, these nucleases may be used for specific modifications of the native genes in the genomes of the recipient organisms. In particular, advances have been made in native CRISPR/Cas system and helped improve its applications in plants. One particular benefit of the CRISPR/Cas9 technology in plants is the relatively low frequencies of off-target mutations in plants compared to the frequency in animals [131, 132]. For example, only 1.6% off-target effects were predicted in rice [133], and no off-target mutations were observed in A. thaliana, Nicotiana benthamiana, Triticum aestivum, and O. sativa [134-138].
For decades scientist utilized the species Agrobacterium to generate transgenic plants. This process was complex and involved insertion of the gene of interest into the transfer DNA (T-DNA) region of large tumor-inducing plasmids. The effort to introduce the gene of interest into T-DNA for subsequent transfer to plants was a challenge because these plasmids are large, in low copy number, difficult to isolate and manipulate in vitro, and do not replicate in Escherichia coli - the preferred host for genetic manipulation. In addition, T-DNA regions from wild-type tumor-inducing plasmids do not contain unique restriction endonuclease sites which are important in the cloning design of a gene of interest. A breakthrough in facilitating this process occurred with the discovery that the T-DNA region and the virulence (vir) genes that are required for T-DNA processing and transfer could be split into two replicons that were introduced into the same cell, thus proteins encoded by the vir genes could act upon the T-DNA in trans to mediate its processing and export to plant. This breakthrough led to the development of the T-DNA Binary vector system [139]. Figure 5 shows the co-integration/exchange systems compared with the T-DNA binary vector systems. Vir proteins encoded by genes on a separate replicon mediate the T-DNA processing from the binary vector and T-DNA transfer from the bacterium to the host cell [2]. Soon after this discovery, the binary systems helped facilitate the ease in cloning into small T-DNA regions, and both T-DNA binary vectors and disarmed Agrobacterium strains harboring vir helper plasmids become more suited for specialized purposes. The common features of binary vectors that are used in genetic engineering experiments include the following: (1) T-DNA left and right border repeat sequences that define and delimit T-DNA. T-DNA border repeat sequences (T-DNA borders) contain 25 bp, (2) A plant-active selectable marker gene,usually for antibiotic or herbicide resistance, or metabolic markers [140], (3) Restriction endonuclease, rare-cutting, or homing endonuclease sites within T-DNA into which the gene of interest is targeted [141, 142], (4) Origin(s) of replication to allow maintenance in E. coli and Agrobacterium, and (5) Antibiotic-resistance genes within the chromosome and within backbone sequences for selection of the binary vector in E. coli and Agrobacterium.
The Binary vector system has the advantage of rapid initial testing of the CRISPR/Cas system. A vector containing different gRNA can be used for transformation of the plant which is already expressing the Cas9 protein. One additional advantage is the inclusion of different combinations of Cas proteins for specific gRNA, which provides for more flexibility in designing the experiment with more targeted efficiency.
The binary vector system has evolved now into a single vector with dual promoters that encode the Cas protein and gRNA. In most of the single vector systems, RNA polymerase II-based promoters such as CaMV35S, ubiquitin are used for the expression of Cas9 gene, and RNA polymerase III-based promoters such as U6, U3 are used for the expression of the gRNA [4]. This system helps reduce the vector size which ultimately results in increased transformation efficiency and gene editing success [143].
This article was updated in August 2016 by Dr. Georgeta Basturea, in March 2019 by Dr. Konstantin Yakimchuk. Dr. Goldi Kozloski added the section "Plant-based genome editing" and updated the article in August 2019. The title of this article was changed from "Genomic Engineering" to "CRISPR and Genomic Engineering" in Oct 2019.
- Moore J, Haber J. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164-73 pubmed
- Epinat J, Arnould S, Chames P, Rochaix P, Desfontaines D, Puzin C, et al. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res. 2003;31:2952-62 pubmed
- Mandell J, Barbas C. Zinc Finger Tools: custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res. 2006;34:W516-23 pubmed
- Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169:5429-33 pubmed
- Jansen R, Embden J, Gaastra W, Schouls L. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43:1565-75 pubmed
- Makarova K, Grishin N, Shabalina S, Wolf Y, Koonin E. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1:7 pubmed
- Jan J, Good W, Freeman R, Espezel H. Eye-poking. Dev Med Child Neurol. 1994;36:321-5 pubmed
- Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109:E2579-86 pubmed
- Chevalier B, Stoddard B. Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001;29:3757-74 pubmed
- Seligman L, Chisholm K, Chevalier B, Chadsey M, Edwards S, Savage J, et al. Mutations altering the cleavage specificity of a homing endonuclease. Nucleic Acids Res. 2002;30:3870-9 pubmed
- Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, et al. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther. 2011;11:11-27 pubmed
- Hoekema A, Hirsch PR, Hooykaas PJJ, Schilperoort RA. A binary plant vector strategy based on separation of vir- and T-region of the Agrobacterium tumefaciens Ti-plasmid. Nature. 1983;303:179-80. Available from: doi.org/10.1038/303179a0
- Todd R, Tague BW. Phosphomannose isomerase: A versatile selectable marker forArabidopsis thaliana germ-line transformation. Plant Molecular Biology Reporter. 2012;19:307-19. Available from: doi.org/10.1007/BF02772829
- Waters V, Hirata K, Pansegrau W, Lanka E, Guiney D. Sequence identity in the nick regions of IncP plasmid transfer origins and T-DNA borders of Agrobacterium Ti plasmids. Proc Natl Acad Sci U S A. 1991;88:1456-60 pubmed
- Wang K, Stachel S, Timmerman B, Van Montagu M, Zambryski P. Site-Specific Nick in the T-DNA Border Sequence as a Result of Agrobacterium vir Gene Expression. Science. 1987;235:587-91 pubmed
- Materials and Methods [ISSN : 2329-5139] is a unique online journal with regularly updated review articles on laboratory materials and methods. If you are interested in contributing a manuscript or suggesting a topic, please leave us feedback.
- reagentmethod
- Adeno-Associated Viral-Mediated Gene Transfer
- Adenoviral Vectors
- Base Editing
- Cloning and Expression Vectors, and cDNA and microRNA Clones Companies
- Current Approaches in C. elegans Research
- DNA Extraction and Purification
- DNA Methylation
- Gene Knockdown Companies
- Killifish
- Venereal Diseases
- Zebrafish Research Methods