Cells create discrete chemical environments in order to organize complex biochemical reactions in space. Such local environments can be separated either by membrane structures or within non-membranous conglomerates. Membrane enclosed compartments like the nucleus, mitochondria, endoplasmic reticulum or lysosomes are well described and are considered as fundamental units of specialized cellular activity: DNA replication and gene expression [3], ATP synthesis [4], protein folding, modification, and transport [5] or degradation [6]. In contrast, other compartments lack membranes. Such compartments have been noticed in cells early on [7, 8], but only during the last decade have been more carefully characterized [9, 10].
These membrane-less compartments, often named “biomolecular condensates” (BC), are mainly composed of proteins and RNA [11, 12], show liquid-like properties, and separate from the rest of cytoplasm/nucleoplasm via a process known as liquid-liquid phase separation (LLPS) [10, 13, 14]. Such membrane-less organelles can form either in the nucleus or in the cytoplasm. Among the nuclear structures, nucleoli – a multifunctional organelle [15, 16], centrosomes – the site of microtubule nucleation [17, 18], Cajal bodies – the home of spliceosomes [19], stress granules [20], and anisosomes [21], are known examples [12]. King MR and Petry S et al investigated the involvement of TPX2 in microtubule nucleation through LLPS [22]. Gibson BA et al provided evidence to indicate that chromatin could undergo LLPS [23]. Wan L et al reported that somatic mutations of ENL, a chromatin reader of acetylated histones, induced self-association through LLPS and contributed to the tumorigenesis of Wilms tumor [24]. In the cytoplasm, ribonucleoprotein complexes associated into P-bodies [25] and stress granules [20] can be found. In addition to P-bodies and stress granules, neuronal cytoplasm also contains another common BC, neuronal transport granules [26]. Yasuda S et al discovered that proteasomes underwent phase separation under stress or ubiquitylation in cytoplasm and nucleoplasm [27]. Artificial protein constructs, for example, ArtiGPUM can be engineered to enable LLPS [28].
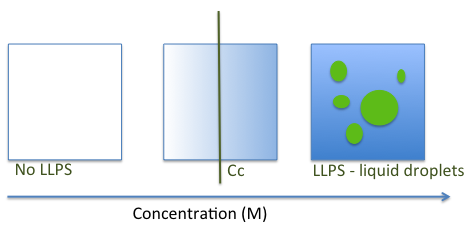
The LLPS process is driven by multiple inter and intra-molecular interactions [12, 29, 30]. An exchange of macromolecule-water interactions for macromolecule-macromolecule and water-water interactions takes place to generate dense phase condensates similar to liquid droplets [23, 29, 30]. Droplet formation depends on the local concentration of biomolecules as well as on variations in environmental conditions, including temperature, pH, salts and presence of other macromolecules [20, 23, 31-33] (Figure 1).
The type of material state of the LLP-separated condensates is defined by the organization of its molecules. They can be liquid-like, gel-like or even solid-like assemblies or can transition through a continuum of states while the cells undergo biological changes like aging or degeneration [34, 35]. Liquid droplets are characterized by short-range molecular order only and have liquid-like properties, like diffusion and surface tension [36]. In these structures molecules arrange in a few layers in an organized fashion, but the next few layers show increased variation in orientation and inter-molecular distances [37]. Hydrogels often form by aggregation of amyloid-like fibers and have less flexible organizations. These structures can be protein sequence dependent and be functional under particular conditions [38, 39]. The weak intermolecular forces in liquid-like assemblies can be disrupted by 1,6-hexanediol [40, 41].
The cell’s advantages of forming these condensates are multiple. A recent review by Banani et al discusses them in detail [12]. Briefly, the distinct properties of biomolecular condensates modulate their functions. Condensates can increase the local concentration of their molecular components, thus increasing the probability of interactions and acceleration of chemical reactions within the structure if all the reaction components are condensed together [14, 42-44]. However, they can affect the enzymatic activity of some enzymes by lowering their specific activity [45] or by inducing changes in the allosteric regulation for others [46]. By grouping together specific enzymes and substrates, condensates can influence the metabolic pathways. For example, concentration of kinases and substrates in the absence of phosphatases was shown to promote specific signalling cascades [47]. Also, by sequestering certain molecules they can limit the availability of substrates or catalysts required by any reactions outside of the condensates [48]. Since they are in a physical state close to the solubility limit of their component macromolecules, the bio-condensates can transition between state phases as a response to minimal environmental changes like concentration, salt, pH or temperature [20, 32, 49]. Such quick changes can be translated into cellular signal transduction pathways that can lead to cellular response to stress [20] or induce pathological states [50]. Furthermore, Klosin A et al demonstrated that phase separation could smoothen the fluctuation of protein concentrations [51]. JZ Zhang et al demonstrated that the diffusible and universal secondary messenger cAMP is sequestered in BC and LLPS is a principal organizer of signaling compartments [41].
Recent cell-free experiments with proteins that contain unstructured domains prone to aggregation have provided hints for phase change behavior in molecular condensates. For example, proteins like FUS and RMB14 form liquid-like conglomerates but with time they become less dynamic and behave more like solids [52, 53]. This behavior is enhanced by particular mutations associated with disease [52] and can disturb the normal gene expression pattern by trapping molecules essential for gene regulation [54, 55] thus contributing to disease.
It is not clearly understood if the ability to form LLPSs is an intrinsic property of all proteins, similar to amyloid-formation, or if the presence of certain protein sequences induces the phase separation under certain conditions [56]. However, before any attempts at studying LLPS/BCs in vitro, a careful analysis of the amino-acid sequences of the studied proteins is recommended [57]. Multiple sequence analysis tools like UNIPROT (https://www.uniprot.org/) [58, 59], BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins), ProtParam (https://web.expasy.org/protparam/) and CIDER (http:// http://pappulab.wustl.edu/CIDER/) [60] are freely available online. Since disordered proteins have a tendency to aggregate, searching intrinsically disordered regions (IDRs), which include sequences that are less likely to form structured domains is a good start [61-63]. Disorder prediction tools like PONDR (http://www.pondr.com/) [41], MobiDB (https://mobidb.mobi/) [64, 65], D2P2 (http://d2p2.pro/) [66] and DisMeta (http://www-nmr.cabm.rutgers.edu/bioinformatics/disorder/) [67] have been recommended [57]. Consulting a specialized database, like DisBind (http://biophy.dzu.edu.cn/DisBind) that contains a collection of experimentally verified binding sites in IDRs, might also be useful [68]. In addition phase-separation (Pi-Pi predictor) [69], prion-like domains (PLAAC - http://plaac.wi.mit.edu/) [70] or fibril forming domains (ZipperDB - https://services.mbi.ucla.edu/zipperdb/) predictors exist.
In vitro model systems employing purified protein components are useful tools when trying to understand the molecular mechanisms that drive phase separation [71]. However, proteins prone to LLPS are rich in unstructured regions and many times prove difficult to purify. When purified proteins were tested for their ability to phase separate and form liquid droplets, the results were often inconsistent [71]. In an attempt to standardize procedures for protein purification and phase separation assays, Alberti et al [71] provides detailed protocols and procedures. Even though protein expression and purification in bacteria is relatively easy, the procedure does not account for the potential role of post-translational modifications in proteins involved in LLPS [72]. Alberti et al (2018) provide details for protein expression in insect cells [71] and describe successful purification procedures for several proteins: P granule and centrosome proteins from C. elegans as well as prion and prion-like proteins from yeast and humans [71]. Both groups emphasize the importance of tag removal as protein tags may affect the behaviour of the proteins in solution. The availability of high-quality purified protein allows investigators to test if a protein condensate can form via LLPS. In addition, mutational analysis makes possible the identification of the specific interacting surfaces involved. Moreover, the requirement for additional molecules, like RNA or different proteins, for the formation of the condensates can be tested. All these are possible when phase-separation assays are performed. Table 1 describes different methods for in vitro LLPS with purified proteins.
| Method of LLPS induction | Procedure | Problems and troubleshooting | Reference |
|---|---|---|---|
| Ionic strength decrease | Concentrated proteins dissolved in high-salt buffer are mixed with low-salt buffer to adjust to physiological salt conditions (150 mM NaCl or KCl) | LLPS do not form if final concentration of protein is too low or if the protein is not homogeneous. | [40, 71] |
| Use of precipitator | Molecular-crowding agents like dextran or PEG-800 are added to the buffer. | Multiple precipitators at different concentrations might have to be tested. | [72, 73] |
| Protein-tag removal | Proteins may be expressed with large tags to improve solubility. Removal of such tags by proteases improves LLPS. | Tag removal might have adverse effects on LLPSs. | [74, 75] |
| Additional biomolecule | Specific binding partners (protein, nucleic acids) can be added to induce LLPS to protein mixtures that do not separate otherwise. | Specific partner might not be known. | [76, 77] |
FRET is a technique commonly used to study interactions between and within molecules, as well as conformational changes within macromolecules [78, 79]. The technique involves the measuring of energy transfer between a donor and an acceptor fluorophore as an indicator of distance between the two molecular components. This method was successfully applied to the study of the nucleolar protein nucleophosmin (NPM1) phase separation [40, 80, 81]. In their studies, Mitrea et al used low concentrations of fluorescently labeled protein within a high concentration of unlabeled protein to eliminate potential artifacts [80] as they monitor continuous fluorescent “waves” generated by slow molecular diffusion, instead of the conventional fluorescent bursts. They demonstrate the mechanisms through which conformational changes within NPM1 control valency and LLPS formation [81].
Fluorescence correlation spectroscopy (FCS) is used to measure the fluctuations of fluorescence intensity from molecules within very small volumes. Such measurements are good indicators of molecular diffusion and have been used for both in vitro and in situ experiments to monitor inter- and intra-molecular interactions [40, 82]. FCS was used to study the movement of the RNA-binding prion-like protein FUS in the nucleus and cytoplasm [33]. The technique allowed the authors to draw the conclusion that RNA-binding triggers the phase-separation behaviour of this protein [33].

As most other techniques, FCS is prone to introducing artifacts [83]. In the case of biological condensate studies, the artifacts in FCS measurements are mostly caused by the differences in refractive indexes of the light in phases of different densities. Modifications of FCS, like two-focus FCS [84, 85] and Z-scan ultra fast scanning FCS (usFCS) [86, 87], were developed to include internal calibrations, and thus reduce the artefacts. Wei et al used the ultrafast-scanning fluorescence correlation spectroscopy to determine the protein concentration within LAF-1 droplets [87].

Once proteins in solution undergo phase-separation, liquid droplets form and the solution changes form clear to turbid. Liquid droplets can be analysed by microscopic examination. Classical light microscopy and its closely related techniques (DIC - differential interference contrast and fluorescence-detection microscopy) provide information on a length scale from 10−7 to 10−3 m and can be used to visualize liquid droplets formed in vitro [27] as well as membrane-less organelles in fixed [88, 89] or living cells [90]. Fluorescence-detection microscopy is often used to detect the distribution of untagged proteins following labelling with fluorophore-conjugated antibodies (immunofluorescence [77] ) or of proteins tagged with a fluorescent protein, such as green fluorescent protein (GFP) (Figure 2). Two variations of this technique are commonly used: wide-field or confocal fluorescence microscopy, both recently reviewed [91, 92]. Because the fixation technique can alter the accessibility to epitopes, the use of fluorescent-fusion tags is better for visualization of molecules inside the cell. To monitor microscopic structures without altering any inter- or intra-molecular interactions, a contrast-based method, such as DIC is advisable. This method exploits the differences in light diffraction by different parts of a sample and provides 3D-images of the sample (Figure 3) [93-97]. DIC is a great method for resolving cellular and in vitro formed boundaries [93, 96, 97].
Electron microscopy is another microscopy technique that can be used to visualize ordered structures, including membrane-less organelles [98]. EM involves electron beams that illuminate fixed-samples and provide images with high resolution (less than 1 A). Transmission electron microscopy (TEM) uses samples coated with heavy metals that provide contrast for visualising structural details of such organelles in vivo [99, 100] or in vitro [101], in fixed and non-fixed cells [99]. Liquid-phase transmission electron microscopy (TEM) is a new tool that allows the direct observation of chemical reactions that take place in solution [102]. By using this technique, Le Ferrand et al [103] captured the nucleation and initial growth steps of LLPS and visualized proteins concentrated into micro-droplets.
| Technique | Advantages | Disadvantages |
|---|---|---|
| Fluorescence-detection microscopy | Stable intermolecular interactions; easy use of immunofluorescence; Use of fluorescent protein fusions allows real-time study of intracellular molecular dynamics | Fluorophores can disturb natural molecular interactions; possible photo toxicity; protein over expression related artefacts. |
| DIC | No molecular disruption caused by added molecules. Well resolved organelle boundaries | Not useful for visualisation of organelle’s components. |
| TEM | High resolution; high contrast | Samples are processed and fixed before analysis. Radiation can damage samples; uneven specimen labelling |
Fluorescence recovery after photobleaching (FRAP) is a fluorescence microscopy technique that can be used to determine the molecular dynamics and mobility of molecules within cells, including within membrane-less compartments formed by liquid-liquid phase separation (LLPS) [21, 27] or in an in vitro droplet assay [104]. In this method protein-bound fluorophores, or other moieties like complemented GFP11 [41], are subjected to light of very high intensity. As a consequence they lose their ability to emit fluorescence, a phenomenon called photobleaching. By observing a recovery of fluorescence at the site of photobleaching, the dynamics of molecular components of any studied system can be monitored. The time course of this recovery can offer information about the diffusional behavior of components of phase-separated membrane-less compartments. In particular, the fluorescence recovery is quick if the phase-separated structures are liquid-like, whereas it is slower in gel-like structures and fails to recover in protein aggregates.
Here we present some recent studies as examples of FRAP use as the technique of choice to study biomolecular condensates in living cells or LLPSs in solution. Gibson BA et al examined the phase-separated reconstituted nucleosomes with FRAP [23]. Frottin F et al measured FRAP of GFP-fusion proteins in HEK293T cells to establish nucleolus as a phase-separated protein quality control entity [105]. Sabari et al used the procedure to study the dynamics of super-enhancers (SE) enriched transcription co-activators BRD4 and MED 1 in living cells [106]. By using the same method, Roubina et al detected that a member of the CBX protein family called Polycomb repressive complex 1 (PRC1) protein chromobox 2 (CBX2) underwent phase separation to form condensates with liquid-like properties [107].
Molecular structures of 10-103 nm in diameter can be detected by monitoring the light scatter, for example, through differential dynamic microscopy [108]. A few variations of light scattering methods like dynamic light scattering (DLS), static light scattering (STS) and small-angle light scattering (SLS) are used to determine the size and shape of particles. LLPSs can be detected by these techniques. DLS allows the detection of large oligomers as well as the determination of the saturation concentration for phase separation of proteins [14, 109]. By determining the onset of scattering in a series of protein dilutions, Thurston GM determined the minimal protein concentration required for the initiation of liquid droplets formation [110]. Moreover, the onset of scattering was monitored as a function of temperature for a constant concentration of protein [111].
Small-angle X-ray scattering (SAXS) is useful for the analysis of intrinsically disordered proteins [112], conformational changes or molecular interactions in solutions [38].
The physical properties of droplets, like viscosity or surface, as well as their dynamics are important as they can influence the biological functions of the organelles. For example, such structures can be more or less viscous - they can behave like solids, gels or liquids. A protein-only droplet can change its properties upon addition of other molecules, e.g RNA, salts [113]. This addition can either fluidize or solidify the droplets [33, 114]. Any such change in vivo can associate with disease [33, 115].
LLPSs are generally more viscous that their surrounding molecules. To determine their viscosity, the diffusion coefficient of a fluorescent marker is being determined by FRAP (see above). The viscosity (η) is then calculated by using a mathematical formula (the Strokes-Einstein equation):
D = kBT / 6πηR
where, D – is the diffusion coefficient, kB – Boltzmann constant, T - temperature (K), and R – the radius of the diffusing particle [13].
The surface tension (Υ), a measure of all interactions that induce minimization of surface area and causes the spherical shape of the droplets, can also be determined experimentally by using either optical traps [52, 116] or sheer stress [13]. When any of these methods are used the inverse capillary velocity (the ratio between viscosity (η) and surface tension (Υ)) can be determined, allowing for the calculation of Υ. The ratio can be obtained by recording fusing droplets and measuring the time it takes for their complete fusion [113]. Other methods for measuring the surface tension are described in Mitrea et al [89] and Alberti et al [57].
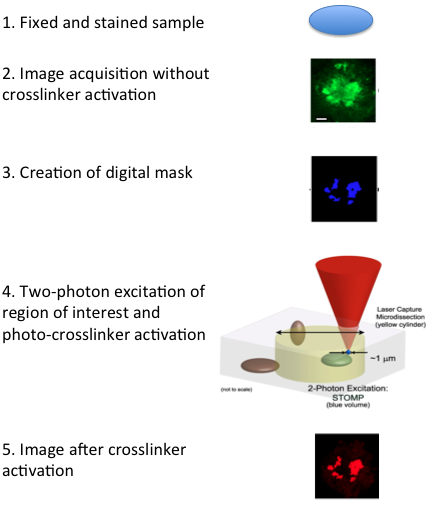
Biocondensates are fluid entities that assemble and disassemble continuously, making their isolation challenging. A novel proteomics technique named spatially targeted optical microproteomics (STOMP), generally used to investigate the protein composition of macromolecular structures of interest without genetic manipulation, can be used to isolate such structures. In this technique, laser light from a microscope is used to image samples that are pre-stained with fluorescent markers while affinity purification tags are photo-crosslinked to protein components within the structure of interest. Following crosslinking, the sample is solubilized and the photo-tagged proteins are isolated and gel separated or identified by mass spectrometry (Figure 4) [1].
Classic fluorescence activated cell sorting (FACS) analysis can be adapted for the sorting of membrane-less organelles from cells. Hubstenberg et al offers an example of such an adaptation. The authors developed a fluorescence-activated particle sorting (FAPS) method in which P-bodies were fluorescently labelled by expressing a P-body marker and were purified from human epithelial cells. In addition, they identified the proteins and mRNAs involved in creating the P-body RNPs, biological condensates that separate repressed mRNAs with regulatory functions [2].
In this technique, fluorophore-bound antibodies are used to visualize the location of specific antigens. The method is widely used in biology and its efficacy depends on the nature of the antigen, quality of the antibody ease of detection of the fluorophore, permeabilization and fixation technique of the sample, and fluorescence imaging of the cell [117-119]. Antibodies specific to proteins within liquid droplets or cellular membrane-less compartments can be used to identify and visualise the organelles by immunofluorescence. Analyses of stress granules in chicken embryo fibroblasts [120] and in mammalian cells are examples of immunofluorescence-based studies of membrane-less organelles [121].
Fluorescence in situ hybridization (FISH) is a frequently used tool proffered for its sensitivity in discriminating multiple targets within the same sample. Most often it is used to detect specific DNA and RNA fragments at the cellular and subcellular levels. Because of these advantages FISH was used in combination with immunofluorescence methods to visualize molecular processes in the nucleus [122]. It has been successful in fixed or frozen cells as well as in unfixed cells [123]. Haroon et al provide a detailed description of a protocol, which, in combination with fluorescence-activated cell sorting (FACS), can be used to analyse single cells or other molecular structures of interest, like membrane-less organelles, from various samples [123].
A variation of the method, single-molecule FISH (smFISH) is extremely sensitive for visualizing, profiling, and quantifying multiple transcripts simultaneously in single cells. In smFISH, researchers use multiple fluorescent probes to detect individual RNAs and measure the sum of the fluorescent intensities from the probes that anneal to the same RNA molecule, thus avoiding secondary amplification [124]. Khong and his co-workers (2017, 2018) used multiple single-molecule FISH probes to examine for the recruitment of particular mRNAs in stress granules [125, 126]. Moon et al used multicolour single-molecule tracking, a variation of smFISH, to determine the kinetics of single mRNAs entry into RNP granules during stress [127].
High-throughput mass spectrometry-based proteomics studies can be used to determine the composition of dynamic structures like bio-condensates (LLPSs or membrane-less organelles). In 2002, Andersen et al used such a technique to identify the proteins in the nucleolus [128]. Since then, protocols that combine high-throughput quantitative proteomics, subcellular fractionation protocols and data analysis made possible the detailed characterisation of proteomes of specific subcellular components [129].
Due to the difficulties in the isolation procedures, the proteome of membrane-less cellular components like stress granules cannot be easily analysed, as weakly associated proteins are likely to be lost during the isolation process [130, 131]. A partial protein composition of stress granules from human cells was determined by a combination of cell fractionation, affinity purification, and mass spectrometry [132]. Proteomics methods were used to identify subcellular conglomerates specific to neurological diseases [133]. Many of the identified proteins are also associated with membrane-less organelles [134]. Chemical or UV-crosslinking can be used to increase the retention of weakly bound proteins in the complex. Also, maintaining specific RNA molecules in the complex might increase the protein association to the macromolecular structure [135].
The RNA composition of membrane-less organelles can be determined by using RNA sequencing methods. One example is the analysis of the RNA components of stress granules performed by Khong et al (2017, 2018) [125, 131], who extracted RNA from isolated granules, sequenced the RNA molecules and validated the results using smFISH as an alternative method. Their results suggest that stress granules form by condensation of mRNAs of genes that are not being transcribes or are transcribed poorly [125].

Together with proteomic analysis, the RNA-seq transcriptomics methods allow for the complete description of the protein-protein and protein-nucleic acids interactions that allow the formation and hold together biological condensates like mRNP complexes, P-bodies and stress granules (Figure 5).
Several imaging techniques used in structural biology including X-ray crystallography, NMR and cryo-electron transmission microscopy (cryo-EM) can be used to analyze at high resolution the structure of macromolecular assemblies present in the biomolecular condensates. However all these methods require homogenous samples of highly purified components, and such materials are not easily obtained in the case of biomolecular condensates. In spite of these particular requirements, a few studies proved successful in using these techniques to determine structural features of liquid droplets, hydrogels, membrane-less organelles or their components.
X-ray diffraction structures of hydrogels formed by low complexity protein regions from proteins like FUS, hnRNP A2 or nucleoporins have been successfully obtained [101, 136].
Kato et al used X-ray diffraction and EM studies to show that low complexity (LC) sequences from FUS and hnRNPA2 form hydrogels that are composed of dynamic amyloid-like fibers, thus revealing a function of LC sequences in the organization of membrane-less cellular structures [101]. Moreover, Hughes et al determined the structures of low-complexity domains associated with membrane- membrane-less assemblies at atomic resolution. All sequences revealed the stacking of segments into kinked β sheets that form protofilaments, as their structural feature. Segments of similar sequence are found in many proteins with diverse functions. For example: RNA binders, nuclear pore proteins, and keratins, all proteins known to participate in networks associated with membrane-less structures [137].
Nuclear Magnetic Resonance (NMR) spectroscopy is the method of choice for structure determination of low-complexity protein fragments in solution. A detailed description of NMR-based techniques that can be used for the study of proteins that associate into cellular bio-condensates was recently included in a review by Mitrea et al [89].
Several NMR-base studies present evidence that structural changes in LC domains of certain proteins associate with phase separation. For example, an alpha-helical structure in the C-terminal LC domain of TDP-43, a protein component of RNP-granules, was shown to be essential for phase separation in vitro [138]. In addition the LC-domains of FUS, the nucleolar protein NPM1 and of the Ddx4 RNA helicase were shown to maintain a disordered conformation while present in phase separated structures [80, 139, 140]. For FUS the intrinsically disordered structure of its LC is preserved even after phosphorylation, while disrupting its phase separation, aggregation and toxicity. This observation was also made using NMR spectroscopy [141].
A large number of NMR experiments use solvent exchange rates to gain information about the structural and dynamical properties of biomolecules [89]. Among the monitored isotopes are 1H, 13C, 15N, 19F and 31P [89]. As all experimental techniques, the classical isotope exchange experiments have their limitations. More recent developments called chemical exchange saturation transfer (CEST) allowed the measurement of solvent exchange in 15N-labeled proteins while monitoring a deuterium isotope shift [142]. By using this new technique, Yuwen et al studied a disordered protein domain that loses the ability to phase-separate when a mutation is being introduced even though the wild type and mutant samples display very similar hydrogen exchange rates [142].
Solution NMR spectroscopy has also been used to assess the tendency of monoclonal antibodies to form LLPSs in solution [143]. In their study Kheddo et al used 1H NMR spectroscopy to determine the causing factors of the LLPS behaviour of an IgG1 mAb (mAb5) antibody in solution. They characterized the separated phases and found a modality to reduce the phase-separation [143]. Without doubt, this can have positive consequences for the antibody-based drug production.
Cryo-electron transmission microscopy (cryo-EM) and cryo-electron tomography (cryo-ET) can provide structural information for biomolecules in their cellular environment. The length scale is from Å to nm, a length between the ones covered by light microscopy and X-ray crystallography or NMR. In Cryo-EM images of multiple identical particles are overlapped and computationally combined to obtain a 3D image of the sample particle. In contract, cryo-ET combines multiple images of the same particle, taken from different angles, to obtain the final image. Because cryo-EM requires a homogenous sample, with identical individual particle, it is not very useful for the imaging of biological condensates, except for the analysis of oligomeric structures formed during nucleation events on phase transitions [89]. In contrast, cryo-ET can be used to study gel-like droplets and membrane-less structures [116].
Cryo-electron tomography (cryo-ET) is a label-free imaging technique that provides high-resolution structural information about organelles inside cells in their native conditions (frozen-hydrated state) [144]. Cryo-ET technique does not use the conventional preparation methods such as chemical fixation, staining, and dehydration but uses a process known as vitrification, which is rapid freezing of aqueous protein solution that preserves the delicate spatial organization of soluble components of the cytosol [145]. Since cryo-ET can be used only for samples with a thickness up to 500 nm [145, 146], focused ion beam (FIB) milling has been used to prepare vitreous samples for cryo-ET. The FIB milling in conjugation with cryo-ET has been successfully used for the observation of biomolecular condensates. A study by Bäuerlein et al has recently shown the intracellular interactions and the structure of huntingtin poly-glutamine (Poly-Q) inclusions in mouse neuron and HeLa cells [147].

Total internal reflection fluorescence (TIRF) microscopy is an optical technique that provides a new and direct approach to study molecular mechanisms in a cellular environment. When light passes through two different materials of different refractive indexes, the ratio of refracted to reflected light changes. At a critical angle of incidence all the light is refracted parallel to the boundary between the two media. At this point all light is internally reflected (total internal reflection) and creates a light wave that transmits into the second medium and decays exponentially. In total internal reflection fluorescence fluorophores within ∼100 nm of the sample surface are excited, while other background illumination is excluded (Figure 6) [148]. By using imaging fluorescent molecules such as membrane dyes, GFP-bound proteins, and fluorochrome-conjugated antibodies, TIRF allows the visualization of single molecules in vivo [149]. TIRF is especially useful for studying protein–DNA and protein-protein biochemical interactions [40, 148]. For example, TIRF has been used to detected and tracked biomolecular condensates located at the interface between the T cell and antigen-presenting cell (APC), organized around the transmembrane adaptor protein LAT (Linker for Activation of T cells) as well as to analyze the actin polymerization in a phase-separated cluster of actin signaling proteins [150]. By using the same method, Peskett et al investigated the structural and material properties of huntingtin exon1 assemblies in mammalian cells and in yeast [151] whereas Zhang et al show that RNAs can drive the phase transition of Whi3, an RNA-binding protein with a long polyQ tract that forms highly heterogeneous structures in the cytoplasm of Ashbya [114].
Bracha et al developed an oligomerizing biomimetic system, named "Corelets," that uses photo-oligomerizable seeds to map phase – separated fractions [152]. The system was used to map complete intracellular phase diagrams that indicate the concentrations at which phase separation occurs and the transition mechanisms that occur in a protein-sequence dependent manner [152].
In eukaryotes, specialized compartments perform specific tasks, like synthesis or degradation of macromolecules. Most of these organelles are separated from the surrounding medium by membranes, but membrane-less condensates of specific macromolecules were found to have important, mostly regulatory, roles. Reinkemeier et al designed an artificial, membrane-less organelle for mammalian cells that worked as artificial ribosomes that translate specific mRNAs in response to a specific codon. These ribosomes are trapped into the synthetic organelles and permit the insertion of synthetic, non-canonical, amino acids into the amino-acids sequence of the synthesized protein. The non-conventional amino acid confers new functions to the protein and can be inserted at any site of interest within the protein [153]. This approach opens multiple possibilities in synthetic cell engineering and biomedical research such as the insertion of novel functional and controllable units into mammalian cells.
- Tulsiani D, Touster O. beta-D-Glucuronidases from the preputial gland of the female rat. Methods Enzymol. 1978;50:510-4 pubmed
- Luzio J, Pryor P, Bright N. Lysosomes: fusion and function. Nat Rev Mol Cell Biol. 2007;8:622-32 pubmed
- Wolf N, Priess J, Hirsh D. Segregation of germline granules in early embryos of Caenorhabditis elegans: an electron microscopic analysis. J Embryol Exp Morphol. 1983;73:297-306 pubmed
- Strome S, Wood W. Generation of asymmetry and segregation of germ-line granules in early C. elegans embryos. Cell. 1983;35:15-25 pubmed
- Boisvert F, van Koningsbruggen S, Navascues J, Lamond A. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8:574-85 pubmed
- Li H, Leo C, Zhu J, Wu X, O Neil J, Park E, et al. Sequestration and inhibition of Daxx-mediated transcriptional repression by PML. Mol Cell Biol. 2000;20:1784-96 pubmed
- Heng M, Heng M. Beta-adrenoceptor antagonist-induced psoriasiform eruption. Clinical and pathogenetic aspects. Int J Dermatol. 1988;27:619-27 pubmed
- Babu M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem Soc Trans. 2016;44:1185-1200 pubmed
- Enderlein J, Gregor I, Patra D, Fitter J. Art and artefacts of fluorescence correlation spectroscopy. Curr Pharm Biotechnol. 2004;5:155-61 pubmed
- Dertinger T, Pacheco V, von der Hocht I, Hartmann R, Gregor I, Enderlein J. Two-focus fluorescence correlation spectroscopy: a new tool for accurate and absolute diffusion measurements. Chemphyschem. 2007;8:433-43 pubmed
- Thurston G. Liquid-liquid phase separation and static light scattering of concentrated ternary mixtures of bovine alpha and gammaB crystallins. J Chem Phys. 2006;124:134909 pubmed
- Collier N, Heuser J, Levy M, Schlesinger M. Ultrastructural and biochemical analysis of the stress granule in chicken embryo fibroblasts. J Cell Biol. 1988;106:1131-9 pubmed
- van de Corput M, Grosveld F. Fluorescence in situ hybridization analysis of transcript dynamics in cells. Methods. 2001;25:111-8 pubmed
- Femino A, Fay F, Fogarty K, Singer R. Visualization of single RNA transcripts in situ. Science. 1998;280:585-90 pubmed
- Andersen J, Lyon C, Fox A, Leung A, Lam Y, Steen H, et al. Directed proteomic analysis of the human nucleolus. Curr Biol. 2002;12:1-11 pubmed
- Super D, Cartelli N, Brooks L, Lembo R, Kumar M. A prospective randomized double-blind study to evaluate the effect of dexamethasone in acute laryngotracheitis. J Pediatr. 1989;115:323-9 pubmed
- Marko M, Hsieh C, Schalek R, Frank J, Mannella C. Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nat Methods. 2007;4:215-7 pubmed
- Materials and Methods [ISSN : 2329-5139] is a unique online journal with regularly updated review articles on laboratory materials and methods. If you are interested in contributing a manuscript or suggesting a topic, please leave us feedback.
- method
- Flow Cytometry - A Survey and the Basics
- Flow Cytometry and Cell Sorting: A Practical Guide
- Identification of Ubiquitinated Proteins
- Incorporating Unnatural Amino Acids into Recombinant Proteins in Living Cells
- LncRNA Research Resources
- Low Abundance Proteins
- Mapping Protein-RNA Interactions by CLIP
- NMR Spectroscopy in Drug Discovery and Development
- NMR in Biomedical Research
- Nanodiscs: Membrane Protein Research in Near-Native Conditions
- Protein Companies
- Protein Expression
- Protein Modification
- Protein/Peptide Tags
- Protein Purification
- Protein Quantitation
- Quantitative Bioanalysis of Proteins by Mass Spectrometry
- RNA Extraction
- RNA Imaging
- RNA Modifications
- RNA-seq
- siRNAs and shRNAs: Tools for Protein Knockdown by Gene Silencing