An overview of base editors.
Genomic editing techniques permit the modification of an organism’s genetic information by either adding, removing or altering the DNA at specific locations in the genome. These are powerful technologies and are of great interest for the treatment of single gene or complex human diseases. However, modifying human DNA raises safety and ethical questions. Consequently, most genome editing research is currently done in model cellular systems or model organisms.
The most popular genome editing tools are zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALEs), and the CRISPR-Cas9 systems. All these tools introduce a double-stranded break (DSB) at specific sites in the DNA. The DSBs are repaired through either the error-prone process of nonhomologous end joining (NHEJ) or by homology-directed repair mechanisms [1]. A detailed description of the main gene editing systems currently used and of some successful approaches to targeted genome editing can be found here.
Despite their constant improvement, all these editing tools continue to display several limitations inherent to their mechanisms of action [2]. One of them, off-target mutagenesis, is due to the introduction of mutations caused by DSBs at genomic sites other than the desired target site. Moreover, because some of the introduced DSBs are repaired by NHEJ, deletions or insertions (indels) at the target site are introduced. Other limitations are technical: production of the nuclease can be laborious and delivery to cells can be challenging. These limitations have increased the demand for more precise and efficient methods for genome modification, be it single nucleotide variation, small insertion or deletion or large chromosomal changes. Alternative approaches that do not rely on homology-directed repair (HDR) have started to emerge in the recent years [3-5].
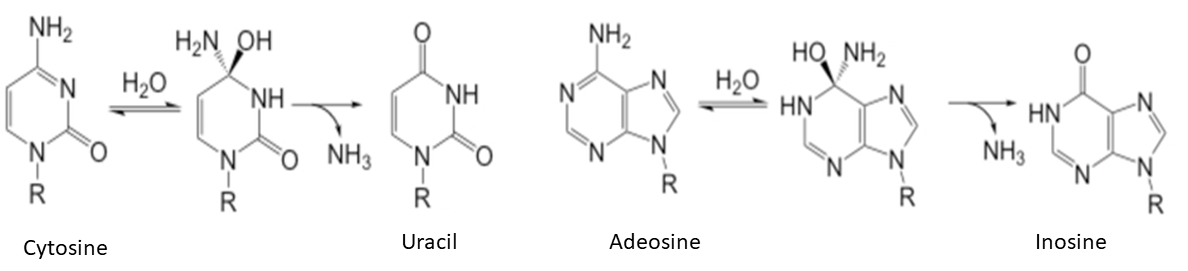
The current alternative to genome editing is base editing (Figure 1). Base editing is the conversion of one target base or base pair into another (e.g. A:T to G:C, C:G to T:A) without requiring the creation and repair of DSBs [3, 4, 6, 7].
The base editing is achieved with the help of DNA and RNA base editors that allow the introduction of point mutations at specific sites, in either DNA or RNA. Generally, DNA base editors consist of a fusion of a catalytically inactive nuclease and a catalytically active base-modification enzyme that acts only on single-stranded DNAs (ssDNAs). RNA base editors are composed of similar, RNA-specific enzymes [7]. Base editing increases the efficiency of gene modification, while reducing the off-target and random mutations in the DNA [3-5].
Base editing is limited to four transition mutations (C→T, G→A, A→G, and T→C) [8]. A new system, prime editing with a prime editing guide RNA (pegRNA) and a Cas9 nickase fused to an engineered M-MLV reverse transcriptase, has been devised to achieve all 12 base-to-base conversions; in addition, it can mediate targeted insertions and deletions [8, 9]. JM Levy et al also devised a split-intein strategy to overcome the limited capacity problem of adeno-associated viral vectors for in vivo base editing [10].
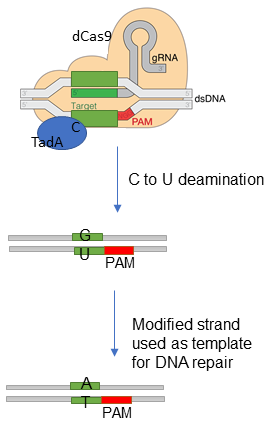
DNA base editors are engineered ribonucleoprotein complexes that act as tools for single base substitution in cells and organism. They were created by fusing an engineered base-modification enzyme [3, 11, 12] and a catalytically deficient Cas9 variant that cannot cut dsDNA, but it is able to unfold the dsDNA in a protospacer adjacent motif (PAM) sequence-dependent manner, such that a guide RNA can find its complementary target to indicate a ssDNA scission site [13]. Cas9 variants with expanded PAMs have been generated [14]. The guide RNA anneals to the complementary DNA, displacing a fragment of ssDNA [15, 16], and directing the Cas9 scissors to the base modification site [4, 5, 17]. The cellular repair machinery will repair the nicked non-edited strand using information from the complementary edited template.
So far, two types of DNA editors, cytosine base (CBEs) and adenine base editors (ABEs) have been developed. They were shown to efficiently and precisely edit point mutations in DNA with minimal off-target DNA editing [18-20]. However, recent findings indicate that off-target modifications are present in DNA [21, 22], and that many off-target modifications are also introduced into RNA by DNA base editors [23, 24].
Cytosine base editors are fusions of cytidine deaminases to CRISPR-Cas nickases. They can efficiently induce C-to-T alterations in DNA [3, 7, 25] by efficiently deaminating the exocyclic amine of the target C to generate U.
The recent report of a first base editor, the cytosine base editor BE1, [3] was rapidly followed by multiple improved versions like BE2, BE3, BE4 and their enhanced variants (the high-fidelity BE3: HF1-BE3, SaBE4, BE4-Gam, SaBE4-Gam, and others). BE1 [3] consists of the rat single-stranded DNA (ssDNA)–specific cytidine deaminase rAPOBEC1 fused to the catalytically dead SpCas9 (dCas9) from Streptococcus pyrogenes via a 16-residue XTEN linker (rAPOBEC1-XTEN-dCas9) [26]. dCas9 is a double point-mutant of the dsDNA-specific nuclease Cas9 that has DNA binding and nickase activity. More specifically, the enzyme contains a point mutation in each of its two catalytic residues: D10A and H840A. The first mutation, D10A, inactivates the enzyme’s dsDNA cleaving function, while the second renders it the ability to nick ssDNA [13].
The dCas9 variant binds at genomic sites of interest, guided by a specific guide RNAs. A protein-RNA-DNA ternary complex is formed in which short (~5-nucleotide) regions upstream of the PAM sequence on the strand noncomplementary to the guide RNA become exposed for modification by the associated cytidine deaminase enzyme. The deaminase acts on any cytidine present in the sequence and hydrolytically transforms them into uracil generating G:U base pairs. BE1 can deaminate cytosines in a variety of different sequence motifs, in vitro [3]. However, inside the cell any G:U base pair is recognized as a mismatch by the cellular repair systems and base excision repair (BER) activity leads to removal of uracils by uracil N-glycosylases (UNGs) rendering the BE1 system unfit for in vivo single base editing [3].
In order to protect the intermediate G:U pair from being excised by uracil-N-glycosylases (UNGs) and to increase the chances of obtaining the desired T:A base pair outcome, the same team added a 83–amino acid uracil glycosylase inhibitor (UGI) directly to the C terminus of catalytically dead Cas9 [27]. The new construct, rAPOBEC1-XTEN-dCas9-UGI, prevented the repair of U:G mismatches back into C:G base pairs and increased the efficiency of base editing in human cells [3]. Based on its ability to convert the C base and protect the resulting U, the new construct increased the maximum base editing yield to 50% and became the second-generation cytosine base editor BE2 [3]. Editing with BE2 generates very few indels (< 0.1%) making it a good choice for applications where indels are not desirable [3].
A third version of the editor, BE3, further increases the editing efficiency of BE2 by manipulating the cellular DNA repair machinery into correcting the non-edited strand. BE3 consists of the same rAPOBEC1-XTEN-dCas9-UGI fusion protein in which the catalytic H840 residue has been restored. Thus, BE3 is rAPOBEC1-XTEN-dCas9(A840H)-UGI, or rAPOBEC1-XTEN-dCas9nickase-UGI, an enzyme unable to cleave dsDNA (Cas9 retains the D10A mutation) but can nick the non-edited strand [3]. This nicking event stimulates the cellular DNA mismatch repair system to remove and resynthesize the non-edited strand [28], now using the information from the already edited complementary strand. BE3 performs more efficient editing than BE2, but at the cost of increasing indel formation at a frequency of up to 1% [28], still much lower than for the classical DSB-mediated approaches [12]. In addition, BE3 generates fewer off-target editing events than Cas9 [3, 20], and this number is further reduced for their high-fidelity variant HF-BE3 [29].
HF-BE3, is an engineered mutant variant of BE3 in which the DNA affinity of dCas9 has been reduced by the introduction of four point mutations (N497A, R661A, Q695A and Q926A) that eliminate nonspecific interactions with the phosphate backbone of the DNA [30] leading to less non-specific editing while maintaining the efficiency of the on-target editing activity [29].
A BE3-like system, called Target-AID, was described independently by the group of Nishida and co-workers [4]. Target-AID consists of an activation-induced cytidine deaminase (AID) from sea lamprey (PmCDA1) fused to Cas9 nickase. The enzyme edits cytosines specifically within the target range of 3-5 bases around the -18 position upstream of the PAM sequence on the DNA strand noncomplementary to the guide RNA in a BE3-like manner. The editing of cytosines is efficient, but similarly to BE3, without high specificity [4].
Kim YB et al developed several enhanced BE3 editors [31]. They identified and addressed the two main limitations of the BE3 editors: the requirement for an NGG PAM sequence that places the target C within a five-nucleotide window near the PAM-distal end of the protospacer, the lack of discrimination between the Cs within that window, The PAM requirement reduces the number of sites in a genome that can be edited by BE3, while the lack of selectivity leads to many unwanted C to T conversions. By using a Cas9 homolog from Staphylococus aureus (SaCas9) [32] and engineered dCas9 variants [33, 34]. Kim YB et al obtained five new editors with distinct PAM sequences (Table 1) that edit at sites not accessible to BE3 [31], thus expanding the number of editable targets. To address the off-target activity of BE3s, the same group developed editors with altered activity windows by introducing specific mutations into the catalytic deaminase (Table 1). Combinations of the two types of constructs led to the development of editors with a narrowed activity window and greater positional selectivity of target sites (e.g. YE1-VQR-BE3, EE-VQR-BE3, YE1-SaKKH-BE3, EE-SaKKH-BE3 -Table1) [31].
Editing efficiency by BE3 can be lower for some target C’s that are immediately downstream of a G [3], depending on the sequence preference of the deaminase APOBEC1. To eliminate the efficiency inconsistencies, Komor AC et al used deaminases with alternative sequence specificities to generate CDA1-BE3, AID-BE3 and APOBEC3G-BE3 [35]. Their analysis revealed that CDA1-BE3 and AID-BE3 edited more efficiently for some target 5′-GC-3′ sequences, while APOBEC3G-BE3 did not show improvements when compared with BE3 [35]. A small-molecule controlled BE3 variant, in which a ligand-responsive self-cleaving catalytic RNA was integrated into the guide RNA, was constructed by Tang W et al [36]. Other base editors recognizing modified PAM (xCas9-BE3 [37] ; SpCas9-NG-BE3 [38] ) were recently developed [39].
Komor et al, (2017) increased the lengths of the existing linkers and added an additional linker followed by a second UGI at the C-terminus of the BE3 construct (Table 1) [35]. Thus, they developed a new improved editor, BE4, with higher efficiency, lower indel rates and decreased incorrect (non-T) product formation than BE3 [35]. Access to different targets was obtained by replacing dCsa9(D10A) with the alternative PAM sequence containing SaCas9 in the SaBE4 variant. To further decrease the indel frequency, the same team fused the DSB-binding bacteriophage Mu protein Gam to the N-terminus of BE4. The role of Gam in this construct is to bind on any double strand free ends, thus inhibiting NHEJ repair and leading these cells to apoptosis. Several BE4 and BE3-Gam fusion constructs were produced (Table 1) and they all exhibited decreased indel frequencies without altered editing efficiency or product purity [35].
CRISPR-Cas9 based editors are not the only base editors developed in the recent years. Yang et al created editors by fusing cytidine deaminases and zinc-finger or TALE-DNA binding domains [40].
| Name | Structural features | PAM | Efficiency and specificity | Refs. |
|---|---|---|---|---|
| BE1 | rAPOBEC1-XTEN-dCas9 | NGG | Apparent editing efficiency: 44% (in vitro), 0.8-7.7% (in vivo) Indel formation: ≤ 0.1% | [3] |
| BE2 | rAPOBEC1-XTEN-dCas9-UGI | NGG | Apparent editing efficiency: 20% (in vivo) Indel formation: ≤ 0.1% | [3] |
| BE3 | rAPOBEC1-XTEN-dCas9(A840H)-UGI | NGG | Apparent editing efficiency: 20-30% (in vitro) 15-75% (in vivo). Indel formation: ≤ 5 % Off-target editing: high genome-wide | [3, 29, 41-43] |
| HF-BE3 | rAPOBEC1-XTEN-dCas9(A840H, N497A, R661A, Q695A and Q926A)-UGI | NGG | Apparent editing efficiency: 30-40% (in vitro) 20-25% (in vivo). Indel formation: ≤ 5 % Off-target editing: high genome-wide | [29, 42, 43] |
| Target-AID | PmCDA1-dCas9-UGI | NGG | Apparent editing efficiency: 3-30% (in vivo) Higher editing efficiency than BE3 when codon optimized for specific organism. Lower indel formation and off-target editing that BE3. | [4, 41] |
| SaBE3 | rAPOBEC1-XTEN-SaCas9(A840H)-UGI | NNGRRT | Apparent editing efficiency: 50-75% (in vivo); Indel formation: ~ 5 % | [31, 32] |
| VQR-BE3 | rAPOBEC1-XTEN-VQR-Cas9(A840H)-UGI | NGAN | Apparent editing efficiency: 10-50% (in vivo) | [31, 33] |
| EQR-BE3 | rAPOBEC1-XTEN-EQR-Cas9(A840H)-UGI | NGAG | Apparent editing efficiency: 10-50% (in vivo) | [31, 33] |
| VRER-BE3 | rAPOBEC1-XTEN-VRER-Cas9(A840H)-UGI | NGCG | Apparent editing efficiency: 10-50% (in vivo) | [31, 33] |
| SaKKH-BE3 | rAPOBEC1-XTEN-SaKKH-Cas9 (A840H)-UGI | NNNRRT | Apparent editing efficiency: >60% (in vivo) | [31, 34] |
| YE1-BE3 | rAPOBEC1(W90Y, R126E)-XTEN-dCas9(A840H)-UGI | NGG | Higher specificity than BE3 | [31] |
| YE2-BE3 | rAPOBEC1(W90Y, R132E)-XTEN-dCas9(A840H)-UGI | NGG | Higher specificity than BE3 | [31] |
| EE-BE3 | rAPOBEC1(R126E, R132E)-XTEN-dCas9(A840H)-UGI | NGG | Higher specificity than BE3 | [31] |
| YEE-BE3 | rAPOBEC1(W90Y, R126E, R132E)-XTEN-dCas9(A840H)-UGI | NGG | Higher specificity than BE3 | [31] |
| YE1-VQR-BE3 | rAPOBEC1(W90Y, R126E)-XTEN-VQR-Cas9(A840H)-UGI | NGAN | Higher specificity than BE3 | [31] |
| EE-VQR-BE3 | rAPOBEC1(R126E, R132E)-XTEN-VQR-Cas9(A840H)-UGI | NGAN | Higher specificity than BE3 | [31] |
| YE1-SaKKH-BE3 | rAPOBEC1(W90Y, R126E)-XTEN-SaKKH-Cas9(A840H)-UGI | NNNRRT | Higher specificity than BE3 | [31] |
| EE-SaKKH-BE3 | rAPOBEC1(R126E, R132E)-XTEN-SaKKH-Cas9(A840H)-UGI | NNNRRT | Higher specificity than BE3 | [31] |
| BE3-Gam | Gam-rAPOBEC1-XTEN-dCas9(A840H)-UGI | NGG | Apparent editing efficiency: < 50% (in vivo); Indel formation: ≤ 5 % Off-target editing: 1-15% | [35] |
| SaBE3-Gam | Gam- rAPOBEC1-XTEN-SaCas9(A840H)-UGI | NNGRRT | Apparent editing efficiency: < 50% (in vivo); Indel formation: ≤ 5 % Off-target editing: 1-15% | [35] |
| xCas9-BE3 (xBE3) | rAPOBEC1-XTEN-xCas9 (A840H)-UGI | NG GAA GAT NGT | Improved DNA specificity; Increased number of SNPs in ClinVar database it can target (~73%). | [37, 44] |
| BE4 | rAPOBEC1-32aa linker-dCas9(A840H) nickase-9aa linker-UGI-9aa linker-UGI | NGG | Apparent editing efficiency: ~50% (in vivo); Indel formation: ≤ 5 % Off-target editing: 1-15% | [35]. |
| SaBE4 | rAPOBEC1-32aa linker-SaKKH-dCas9(A840H) nickase-9aa linker-UGI-9aa linker-UGI | NNNRRT | Apparent editing efficiency: ~50% (in vivo); Indel formation: ≤ 5 % Off-target editing: 1-15% | [35]. |
| BE4-Gam | Gam- rAPOBEC1-32aa linker-dCas9(A840H) nickase-9aa linker-UGI-9aa linker-UGI | NGG | Apparent editing efficiency: ~50% (in vivo); Indel formation: ≤ 1.5 % Off-target editing: 1-15% | [35] |
| SaBE4-Gam | rAPOBEC1-32aa linker-SaKKH-dCas9(A840H) nickase-9aa linker-UGI-9aa linker-UGI | NNGRRT | Apparent editing efficiency: ~50% (in vivo); Indel formation: ≤ 1.5 % Off-target editing: 1-15% | [35] |
| xCas9-BE4 (xBE4) | rAPOBEC1-32aa linker-xCas9(A840H) nickase-9aa linker-UGI-9aa linker-UGI | NG GAA GAT NGT | Improved DNA specificity; Increased number of SNPs in ClinVar database it can target (~73%). | [37, 44] |
Like cytosine, adenine can be deaminated to change its standard pairing. Its deamination yields inosine (Figure 1 B) read as G and paired with C by DNA polymerases. Since there were no known DNA adenine deaminases, Gaudelli et al (2017) created an adenine base editor [5]. Others were developed afterwards [45-47].
Adenine base editors (ABEs) convert an A:T base pairs to a G:C base pairs. They consist of three main components: a mutant transfer RNA adenosine deaminase (TadA), a Cas9 nickase, and a sgRNA. The first ABE was engineered by using directed evolution of the E. coli tRNA adenosine deaminase TadA into a ssDNA binding adenine deaminase [5]. Because the TadA is a dimeric enzyme and a first evolved TadA variant -dCas9 fusion did not edit A to I very efficiently on ssDNA, the team engineered a heterodimeric proteins that included a wild-type non-catalytic TadA monomer, an evolved TadA* monomer and a Cas9 nickase (TadA–TadA*–Cas9 nickase). These constructs greatly increased the adenine base editing efficiency [5]. The most efficient of all is ABE7.10, contains 14 amino-acid substitutions in TadA and edits at As located within a 4-7 bases target window. Three other related constructs ABEs 6.3, 7.8, and 7.9 have slightly larger editing windows at the expense of their editing efficiency (Table 2) [5]. When compared to CBEs, all engineered ABEs are superior regarding product purity, as removal of I from DNA is not a frequent event.
The development of new generations of CBEs, lead to the rapid engineering of their adenosine counterparts. Thus, adenosine specific editors were constructed containing most of the Cas9 variants that target non-canonical PAM sequences (Table 2).
| Name | Structural features | PAM | Efficiency and specificity | Refs. |
|---|---|---|---|---|
| ABE7.9 | TadA-32 aa linker-TadA mutant-32 aa linker-SpCas9(D10A, H840A) | NGG | ~ 50% editing efficiency in human cells 99 % product purity Indel rates < 0.1% | [5] |
| ABE7.10 | TadA-32 aa linker-TadA mutant-32 aa linker-SpCas9(D10A, H840A) | NGG | ~ 50% editing efficiency in human cells 99 % product purity Indel rates < 0.1% | [5] |
| VQR-ABE | TadA-32 aa linker-TadA mutant-32 aa linker-VQR-Cas9(A840H) | NGAN | Up to 23% editing efficiency | [47] |
| VRER-ABE | TadA-32 aa linker-TadA mutant-32 aa linker-VRER-Cas9(A840H) | NGCG | Up to 8% editing efficiency | [47] |
| SaKKH-ABE | TadA-32 aa linker-TadA mutant-32 aa linker-SaKKH-Cas9(A840H) | NNNRRT | Increased DNA specificity | [47] |
| NG-ABE | TadA-32 aa linker-TadA mutant-32 aa linker-SpCas9-NG | NG | Up to 32% editing efficiency | [38, 47] |
| ABEmax | bpNLS ABE7.10 | NGG | Indel rates < 1.6% Editing efficiency and product purity comparable with ABE7.10 Increased SNP correction rate (~75%) | [48] |
| xABE | TadA-32 aa linker-TadA mutant-32 aa linker xCas9(A840H) nickase | NG GAA GAT NGT | Improved DNA specificity; 16-70% editing efficiency in human cells Increased number of SNPs in ClinVar database it can target (~71%). | [37] |
Further improved versions of both cytidine (reassembled BE3, BE3RA; BE4max; AncBE4max) and adenine (ABEmax) DNA base editors were obtained by codon optimization and addition of nuclear localization signals [39, 48].
Base editing in RNA has primarily relied on the use of the double-strand RNA specific deaminase (ADAR), an enzyme that converts A to I both at specific and non-specific sites in mRNAs. Targeted activity to desired sites was obtained by antisense-RNA mediated editing. In this technique, specific antisense RNA molecules with variable degrees of homology to the target molecule, are used in conjunction with the ADAR deaminase. The target A is indicated by an A:C mismatch in the mRNA-antisense RNA duplex, and deaminate to I. In order to ensure the colocalization of the two molecules, ADAR is physically linked to the antisense-RNAs.
ADAR-based editors (Table 3) known as Antisense-RNA mediated RNA editing, ADAR-BoxB mediated RNA editing and ADAR2-mediated RNA editing were all developed based on ADAR-RNA fusions. In antisense-RNA editing modified antisense RNAs fused to a SNAP-tagged ADAR deaminase domain enabled editing both in vitro and in vivo [49-51]. ADAR-BoxB mediated RNA editing was based on a fusion between the ADAR deaminase domain and the BoxB RNA-binding λ-phage N protein. This protein fusion was linked to two BoxB hairpin containing antisense-RNA that could localize the deaminase to the target A [52, 53]. The full-length ADAR2 based editing involves an RNA fusion consisting of a 5’ R/G-binding motif hairpin, the native binding sequence for full length ADAR2, and an antisense region complementary to the target RNA [54, 55]. Due to the substrate preference of ADAR proteins, all these editors display limited efficiency for editing in certain sequence contexts. For example, GAN sites are not efficiently edited by any of these editors.
An alternative approach for RNA editing that uses a CRISPR-Cas system was developed by Cox et al, (2017). This group used a catalytically dead RNA-guided Cas13b enzyme (dPspCas13b) to localize the ADAR deaminase to the target RNA [56]. The system was named RNA Editing for Programmable A-to-I Replacement (REPAIR) and uses a Cas13b-hyperactive ADAR2 deaminase domain single mutant (E488Q) fusion along with a guide RNA that leads the enzyme to the target via a 50-nt spacer containing a central A:C mismatch [56]. In contrast to ADAR-based editors, REPAIR can edit adenines with higher efficiency in all sequence context, overcoming the ADAR induced substrate limitations [56]. However, the off-target editing level was not improved until mutations that reduced the non-specific RNA binding affinity of ADAR2 were introduced in the deaminase domain of ADAR2 (E488Q). This led to a second variant of REPAIR: REPAIRv2 consisting of protein fusion dPspCas13b- ADAR2DD(E488Q/T375G) and REPAIR guide RNA (see above) [56].
Recently, Abudayyeh et al used directed evolution to select an ADAR2 deaminase domain able to deaminate C. The new system, termed RNA editing for Specific C-to-U Exchange (RESCUE) is based on REPAIR and can be guided to any RNA substrate to perform C-to-U editing, while maintaining its A-to-I editing function [57].
| Name | Structural features | Conversion | References |
|---|---|---|---|
| Antisense-RNA mediated RNA editing | SNAP-ADAR2DD + chemically modified anti-sense RNA | A-to-I | [49-51] |
| ADAR-BoxB mediated RNA editing | λN-ADAR2DD + BoxB hairpin antisense-RNA | A-to-I | [52, 53] |
| Full length ADAR2-mediated RNA editing | ADAR2 + RNA fusion: 5’ R/G-binding motif hairpin- native binding sequence of ADAR2- antisense-RNA | A-to-I | [54, 55] |
| REPAIRv1 | dPspCas13b- ADAR2DD(E488Q) + gRNA with central A:C mismatch | A-to-I | [56] |
| REPAIRv2 | dPspCas13b- ADAR2DD(E488Q/T375G) + gRNA with central A:C mismatch | A-to-I | [56] |
| RESCUE | dPspCas13b- evolvedADAR2DD(E488Q/T375G) + gRNA with central A:C mismatch | C-to-U A-to-I | [57] |
Base editing technology is a great tool for making single-base corrections in genomes, without a high risk of gene disruption by insertion or deletion [3, 4, 6, 7]. However, several limitations have been identified.
DNA base editing yields a lower rate of indel formation when compared with genome editing, but indels are still detectable [3, 5, 58]. Generally, the frequency of introduced indels is lower for ABEs than for CBEs [5]. Komor et al indicated that the number of indels can be greatly reduced by fusing the bacteriophage Mu-derived Gam (Mu-GAM) protein to BE4 [35] or by fusing a SunTag to the N-terminus of the Cas9(D10A) nickase [59].
Base editors are great tools for introducing point mutations at desired loci without breaking DNA. However, the choice of target bases is limited by the presence of specific protospacer adjacent motif (PAM) sequences that determine the editing frequencies within the genome, as most base editors are based on the CRISPR/Cas9 system [3, 5]. To broaden the window of DNA sequences that can be edited, several groups have created variants of the pioneering BE either by bacterial selection-based or phage-assisted evolution [34, 37, 60], structure-based enzyme mutations [31, 61] or protein engineering [59]. Both CBEs and ABEs have been developed to recognize various PAM motifs [37, 38, 62-66]. SH Chu et al inserted the deaminase domains within the Cas9 proteins - inlaid base editors (IBEs) to expand the canonical editing windows and to reduce off-target editing frequency [67].
Base editors generally edit one or multiple Cs or As located within a larger activity window, dictated by the distancing between the protein domains of the editor. An in depth analysis of on-target and off-target editing by a variety of CBEs and ABE used to generate mutant mice embryos [68] indicated that ABE almost exclusively generates desired A:T to G:C edits, while different generations of CBEs generate mutations at undesirable places in various amounts. Similarly, attempts to edit single-nucleotides without introducing undesirable off-target mutations brought into light the need to optimize the base editing systems [68-70]. In order to obtain high specificity and high efficiency editors, attempts were made to narrow the catalytic window by removing non-essential sequences from the deaminase and testing linkers of various lengths, such that a single base within the catalytic window is edited with high precision and efficiency [71]. In addition, codon optimization techniques and addition of nuclear localization signals led to optimized base editors that efficiently modify base targets in mouse and human cells [39], or in plants [72, 73].
The application of base editing is limited by off-target activity. This occurs when additional Cs or Ac are edited at site proximal to the target base in the same DNA molecule. Off-target DNA base editing can be caused by gRNA–dependent or gRNA–independent editing events.
gRNA dependent events are caused by RNA-guided binding of the Cas9 domain to DNA sites that are homologous to the target DNA sequence [21, 22]. CasOFFinder can often be used to predict the off-target sites, which can be further examined [74, 75]. This type of events has been eliminated by improving the DNA specificity of Cas9 [29] or by adding 5′-guanosine nucleotides to the gRNA [21]. By introducing mutations in BE3, Ress et al [29] increased its DNA specificity and created a high-fidelity base editor HF-BE3, the first editor with reduced off-target activity in human cells. Limited off-target mutations were observed when BE3 was used to obtain sheep with defined point mutations [76]. However, it was recently observed that cytosine base editors BE3 and high-fidelity BE3 (HF1-BE3) induce unexpected and unpredictable genome-wide off-target mutations in mouse embryos [42] and in rice [43]. In contrast to CBEs, ABEs in these studies did not result in significant off-target DNA mutations. These events were attributed to gRNA–independent off-target editing caused by the deaminase binding of C or A bases in a Cas9-independent manner [42, 43]. In order to minimize the off-target mutations, further optimization in the cytidine deaminase domain and/or the UGI component were suggested [43].
Recently published articles from two independent groups suggest that DNA base editing by CBEs causes a high number of unpredictable off-target RNA base changes [23, 24]. In order to eliminate the RNA off-target activity of CBEs, the researchers destabilized the RNA binding capacity of the editor by introducing two point-mutations. The BE3 double mutant (W90Y and R126E) showed reduced RNA off-target effects without diminishing its DNA on-target activity [24]. The same effect was achieved by using a mutant human cytidine-deaminase [24].
ABEs have been reported to exhibit minimal off-target DNA editing [42, 43]. To determine if they display any RNA off-targeting, Rees et al [77] overexpressed the most efficient ABE variant, ABEmax, in human cells and performed transcriptome-wide as well as specific mRNA sequencing to determine if ABEmax induces undesirable A to I events in the transcriptome. They found that ABEmax induces a low-level of non-specific RNA editing. The frequency of such events was greatly reduced, without loss of specific and efficient DNA editing, by introducing a point mutation into TadA (E59A or E59Q) or into the evolved TadA monomer, TadA* (V106W) [77].
During the only 3 years since their invention, BEs have been continuously optimized and adapted for a series of applications in basic research, medicine and agriculture. They are promising tools for many applications. Among these are: single point mutations corrections for diseases, single point mutation generation for the creation of animal models, gene inactivation for phenotypic studies or creation of crops with desirable characteristics.
Correction of pathogenic point mutations is a major application of base editing. BEs work can be applied to correct 61% of human pathogenic mutations listed in the ClinVar database [44]. Genes have already been reverted to wild type in cultured cells or embryos by base-editor mediated point mutation corrections. Examples include the conversion of the cancer-associated p53 mutation Tyr163Cys in breast cancer cells [3], the correction of the Alzheimer’s disease associated allele APOE4 in mouse cells [3], of the MPDUI Leu119Pro mutation in fibroblasts [48, 78] or of the beta-thalassemia HBB mutant allele in human cells [79, 80]. Pathogenic alleles were also corrected in mouse [81] and human embryos [80, 82, 83], after direct injection of BEs’ mRNAs and gRNAs.
By using viral vectors to deliver base editors and their associated gRNAs, mutations could be corrected in animal models as well. Ryu et al delivered trans-splicing adeno-associated viral vectors, to muscle cells in a mouse model of Duchenne muscular dystrophy to correct a nonsense mutation in the Dmd gene [81]. Similarly, Chadwick et al delivered an adenoviral vector encoding for BE3 and a nonsense mutation carrying gRNA into the livers of adult mice to introduce site-specific nonsense mutations into the Pcsk9 gene [84]. Song CQ et al detected the expression of fumarylacetoacetate hydrolase (FAH) in the hepatocytes of FAHmut mice (a model of tyrosinemia) after correcting a point mutation with ABE6.3 or its improved version - RA6.3 adenosine base editor [85].
Making specific base changes in vivo, in cell cultures or animal models, greatly facilitate the study of the effect of single point mutations on certain phenotypes. Since their invention in 2016, BEs have been used to insert point mutations in bacteria, plants and animals. For example, Zhang et al efficiently introduced single-base changes in zebrafish by using base editing. This allowed them to create a zebrafish model for human oculocutaneous albinism, a disease in which point mutations in the tyr gene lead to protein loss of function and a deficiency in pigment formation [86]. Hess et al mutated the target of the cancer therapeutic bortezomib, PSMB5, and identified known and novel mutations that confer bortezomib resistance [87]. Similarly, Ma et al identified new mutations that confer resistance to treatment for myeloid leukemia [88]. BEs have facilitated the faster generation of mouse models of human diseases caused by single-nucleotide substitutions [89].
Single base alterations in promoter regions can be used to control an otherwise constitutive promoter or to repair its correct function. For example, multiple beta-globin gene promoter mutations are known to cause β-thalassemia [90, 91]. Gehrke et al showed that such mutations can be repaired by BEs to restore the correct beta-globin gene expression.
In addition to the work of Ryu et al (see above, [81] ), other groups achieved gene disruption by introducing premature stop codons via a BE system. CRISPR-Stop [92] and iSTOP [93] are two BE-based tools created with this purpose. K Musunuru et al, for example, knocked down PCSK9 through a base editor and lowered cholesterol level in cynomolgus monkeys [94]. A database of gRNAs for iSTOP targeting of several eukaryotic species has been provided by Billion et al [93].
Despite being just recently developed, base editors had already been successfully used in bacteria and animals and proved especially useful for crop alterations as an alternative to the challenging DSB-induced HDR approaches and conventional breeding [95, 96].
BE3s were used to edit genes in rice, wheat and corn with C:G to T:A editing efficiencies of 12-44% [19, 97, 98]. Shimatani et al used the Target-AID system to induce herbicide-resistant point mutations in rice and to improve tomato crops [11, 99]. ABE editing also proved highly efficient in rice [46, 100].
Tang and Liu used base editors to develop CRISPR-mediated analog multi-event recording apparatus (CAMERA) systems to register the duration and order of cellular events in the DNA of bacteria and mammalian cells [101]. The system designed for bacteria, uses an engineered BE2 whose expression can be controlled by antibiotics or other small molecules and a constitutively transcribed guide RNA targeted to a specific region in a recording plasmid. When complexed with BE2, the guide RNA introduces a C:G to T:A mutation at the specific position in the plasmid. The system was multiplexed by using multiple responsive guide RNA expression cassettes and was adapted for use in mammalian cells as well. The system can record, erase and re-record changes in multiple stimuli simultaneously including antibiotics, nutrients, viruses, light and changes in signaling events, by using a less than 100 cells [101].
Base editing in mammalian cells can be performed in the research laboratory by following a few steps [3] :
- Design the guide RNA of targeting the site of interest. Good webtool that helps design, evaluate and clone guide sequences for the CRISPR/Cas9 system are CRISPOR (http://crispor.org) [102], BE-Designer (http://www.rgenome.net/be-designer/) [103].
- Clone the gRNA into the gRNA expression vector compatible for your system. Many gRNA empty vectors have been deposited at Addgene (https://www.addgene.org/crispr/empty-grna-vectors/). Note that the presence of Cas9 protein is also required. Cas can be expressed from the same plasmid with the target-specific gRNA, from a separate plasmid or from a gene pre-inserted into the cells’ genome.
- Extract transfection-quality plasmids for your gRNA and base editor of choice.
- Transfect your cells of interest.
- Grow cells.
- Harvest cells and extract DNA.
- Quantifying base editing efficiency using high-throughput sequencing. Sequencing data can be analyzed by using the free webtool BE-Analyzer (http://www.rgenome.net/be-analyzer) [103].

Genome and base editing tools are very powerful tools that have been constantly improved since the development of the first CRISPR-Cas editing system less than a decade ago [13, 104]. For genome editing the CRISPR system uses a guide RNA attached to a nuclease to produce a double-stranded DNA cleavage at a target site in a genome. The cellular repair systems repair the cut DNA ends, but occasionally insert or delete bases (Figure 3). Base editors use CRISPR components, guide RNAs and a mutant Cas9, but do not cut the DNA. Instead they use deaminase enzymes such as TadA and ADAR enzymes to chemically convert single bases without altering the surrounding sequence. While genome editing is specific to DNA manipulation, base editing can be performed in both DNA and RNA molecules (Figure 3).
Because it eliminates the unwanted insertion and/or deletions caused by gene editing, base editing seems the method of choice for fixing point mutations. However, due to their innate properties, like PAM sequence dependence, base editors cannot act upon all bases in a genome. Thus, many point-mutations associated with disease might still not be correctable by base editing. In addition, the unexpected high number of RNA off-target events introduced by CBEs must be addressed before base editing becomes a tool fit to cure diseases.
- Moussavian S, Becker R, Piepmeyer J, Mezey E, Bozian R. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci. 1985;30:211-4 pubmed
- Singh H, Maheshwari S, Gupta R. Deterioration of tetanus antitoxin at 4-8 degrees C. J Biol Stand. 1982;10:91-4 pubmed
- Mol C, Arvai A, Sanderson R, Slupphaug G, Kavli B, Krokan H, et al. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell. 1995;82:701-8 pubmed
- Montiel González M, Vallecillo Viejo I, Rosenthal J. An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res. 2016;44:e157 pubmed
- Schenk B, Imbach T, Frank C, Grubenmann C, Raymond G, Hurvitz H, et al. MPDU1 mutations underlie a novel human congenital disorder of glycosylation, designated type If. J Clin Invest. 2001;108:1687-95 pubmed
- Eng B, Walker L, Nakamura L, Hoppe C, Azimi M, Lee H, et al. Three new beta-globin gene promoter mutations identified through newborn screening. Hemoglobin. 2007;31:129-34 pubmed
- Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109:E2579-86 pubmed
- Materials and Methods [ISSN : 2329-5139] is a unique online journal with regularly updated review articles on laboratory materials and methods. If you are interested in contributing a manuscript or suggesting a topic, please leave us feedback.